Резюме. На долю острых лейкозов приходится до 30% всех болезней кроветворной и лимфоидной систем у детей. Начальный период лейкоза характеризуется полиморфизмом клинической симптоматики, что затрудняет своевременную диагностику и лечение этой патологии. Причинами обращения больных лейкозом за медицинской помощью, к врачам различных специальностей, являются: наличие лихорадки, геморрагического синдрома, нарастающая слабость, анемия, увеличение лимфатических узлов печени, селезенки, боли в суставах и костях, нарушение стула и др. В статье представлены данные литературы и собственные наблюдения острого лимфобластного лейкоза у детей грудного возраста, госпитализированных в инфекционный стационар с направительными диагнозами: эпидемический паротит; кишечная инфекция – энтерит. Описание клинической симптоматики и течения заболевания представлено в виде анализа клинической ситуации с целью медицинского образования и дифференциальной диагностики с паротитной инфекцией и энтеритом инфекционной этиологии. Проведение анализа клинических симптомов, имевшихся у пациента с направительным диагнозом «паротит», позволяло установить существенные отклонения от обычного течения паротитной инфекции уже при первичном обращении за медицинской помощью. Односторонний паротитный субмаксиллит встречается редко. Чаще поражаются обе железы в комбинации с поражением околоушных желез. Распространенный отек клетчатки более характерен для подчелюстного лимфаденита, а не субмаксиллита, что в сочетании с гепатомегалией свидетельствовало против паротитной инфекции. У ребенка с направительным диагнозом «кишечная инфекция, энтерит» наличие лимфаденопатии, геморрагического синдрома, гепатоспленомегалии при слабо выраженном кишечном синдроме ставило этот диагноз под сомнение. Поражение кишечника при лейкозе встречается часто и обуславливается синдромом энтеропатии вследствие экстрамедуллярного поражения с кровоизлияниями в слизистую оболочку кишечника, активацией условно-патогенных бактерий, входящих в состав микрофлоры кишечника. Причиной ошибок диагностики лейкоза в приведенных наблюдениях являлось отсутствие анализа клинической симптоматики, позволяющего установить несоответствия имеющихся у больного симптомов предполагаемому диагнозу. Односторонний подход к оценке клинических симптомов, имевшихся у больных, отсутствие настороженности врачей в плане заболевания крови явились причинами ошибочной постановки диагноза при первичном обращении пациентов за медицинской помощью.
Острые лейкозы (ОЛ) представляют собой группу заболеваний крови, на долю которых приходится до 30% и более всех болезней кроветворной и лимфоидной систем у детей до 5 лет [1].
Причинами обращения больных лейкозом за медицинской помощью к врачам различных специальностей являются лихорадка, геморрагический синдром, нарастающая слабость, анемия, увеличение лимфатических узлов, печени, селезенки, боли в суставах и костях, нарушения стула и др. [2, 3].
Клиническая картина острого лейкоза складывается из ряда периодов: предлейкозный, острый, ремиссии, рецидив, терминальный [4].
Предлейкозный период характеризуется рядом неспецифических симптомов (повышение температуры тела, вялость, снижение аппетита, бледность кожных покровов), что встречается при различных заболеваниях как инфекционного, так и неинфекционного характера. Более специфичными признаками в предлейкозном периоде являются изменения в общем анализе крови (анемия, гранулоцитопения, тромбоцитопения). Следует отметить, что у 20-30% больных лейкозом эти изменения в гемограмме могут отсутствовать [1].
Симптоматика острого периода лейкоза представлена рядом синдромов:
- интоксикационный – повышение температуры тела до 37,6-38,5 °С и выше, слабость, вялость и др.;
- анемический – головокружение, бледность кожных покровов и слизистых оболочек. Возможны одышка, тахикардия. В общем анализе крови снижается количество эритроцитов, уровень гемоглобина, цветовой показатель;
- геморрагический – кровоизлияния различной локализации в виде петехий, экхимозов на коже и в подкожно-жировой клетчатке. Кровотечения как следствие тромбоцитопении;
- гиперпластический – увеличение различных групп лимфатических узлов (чаще подчелюстной и околоушной области). Консистенция лимфоузлов плотноэластическая, болезненность при пальпации отсутствует. Кожа над лимфоузлами не изменена. Увеличиваются печень и селезенка;
- костно-суставной синдром (встречается только у части больных) – боли в суставах и костях, усиливающиеся в ночное время.
Возможны экстрамедуллярные поражения – центральной нервной системы (нейролейкемия), легких, половых органов, а также энтеропатии вследствие лейкозной инфильтрации с кровоизлияниями в слизистую оболочку кишечника, что клинически сопровождается изменениями консистенции стула, его частоты, наличием примесей крови [5-7].
В остром периоде лейкоза в гемограмме уменьшается количество эритроцитов, снижается уровень гемоглобина, число лейкоцитов увеличено (гиперлейкоцитоз) или снижено до лейкопении, определяются бластные клетки [8, 9].
В терминальном периоде лейкоза изменения в периферической крови нарастают (анемия, гранулоцитопения, тромбоцитопения), увеличивается количество бластных клеток [10, 11].
Приводим собственные клинические наблюдения.
Клиническое наблюдение № 1
Пациент О., 10 месяцев, от первой беременности, первых срочных родов. Масса тела при рождении – 3400 г. Оценка по шкале Апгар – 8 баллов. С 3 месяцев – на смешанном вскармливании. Рос и развивался удовлетворительно. В 4 месяца перенес ОРВИ. Лечение проводилось по месту жительства.
Настоящее заболевание началось 8.03.2018 г. с повышения температуры тела до 37,5-38 °С. Ребенок стал вялым, отказывался от еды. 11.03.2018 г. (на 4-й день от начала болезни) появилось опухолевидное образование в области шеи справа, что послужило основанием для госпитализации в инфекционную больницу с направительным диагнозом: «Паротитная инфекция».
Эпидемиологический анамнез: контакт с инфекционными больными отсутствует. Вакцинирован по возрасту. Аллергический и наследственный анамнез не отягощен.
При осмотре 11.03.2018 г. – состояние средней тяжести. Температура тела – 37,5 °С. Лимфаденопатия. В подчелюстной области справа – опухолевидное образование до 3,5 см в диаметре, плотноэластической консистенции, безболезненное при пальпации, с умеренным отеком тканей, распространяющимся на лицо и шею.
Число дыхательных движений (ЧДД) – 32 в минуту. Дыхание пуэрильное, проводится по всем полям, хрипов нет. Число сердечных сокращений (ЧСС) – 128 ударов в минуту. Тоны сердца умеренно приглушены, шумов нет. Слизистая оболочка полости рта влажная, бледно-розового цвета. Изменения в области выводного протока слюнной железы отсутствуют. Зев бледно-розового цвета. Живот мягкий, безболезненный при пальпации. Печень выступает на 4 см за край реберной дуги (по среднеключичной линии). Селезенка не пальпируется. Стул – кашеобразный, без видимых примесей. Мочится удовлетворительно. Цвет мочи – соломенно-желтый.
В общем анализе крови от 11.03.2018 г.: эритроциты – 3,2 × 1012/л, гемоглобин – 96 г/л, лейкоциты – 18,9 × 109/л, нейтрофилы – 22%, лимфоциты – 72%, тромбоциты – 189 × 109/л, ретикулоциты – 0,5%, скорость оседания эритроцитов (СОЭ) – 12 мм/час. Биохимический анализ крови: общий белок – 60 г/л, общий билирубин – 20 мкмоль/л (прямой – 5, непрямой – 15 мкмоль/л).
Предварительный диагноз: «Паротитная инфекция. Субмаксиллит».
В течение двух последующих дней сохранялась субфеб-рильная температура, появилась бледность кожных покровов. Локальные изменения в подчелюстной области справа сохранялись. Селезенка увеличилась и выступала из-под края реберной дуги на 2 см.
Повторное исследование общего анализа крови 14.03.2018 г. (7-й день от начала болезни) продемонстрировало снижение количества эритроцитов до 2,1 × 1012/л, показателя гемоглобина – до 70 г/л, увеличение количества лейкоцитов до 25,6 × 109/л, лимфоцитов – до 86%, а также тромбоцитопению – 160 × 109/л, наличие бластных клеток, что явилось основанием для перевода пациента в гематологическое отделение. Проведенное в последующем исследование миелограммы выявило бластную трансформацию (22% бластных клеток), а иммунофенотипирование бластной популяции соответствовало B-клеточному варианту острого лимфобластного лейкоза. По результатам цитохимического исследования костного мозга реакция бластных клеток на гликоген была положительная, на липиды и пероксидазу – отрицательная.
В приведенном наблюдении можно увидеть существенные отклонения от обычного течения паротитной инфекции, которые не были учтены врачами поликлиники и приемного отделения при поступлении больного в стационар. Односторонний паротитный субмаксиллит встречается редко. Обычно поражаются обе железы в комбинации с поражением околоушных желез. Распространенный отек клетчатки не характерен для субмаксиллита, что требовало исключения у пациента подчелюстного лимфаденита. Не нашла должной оценки и гепатомегалия, которая не характерна для паротитной инфекции. Считается, что шейный лимфаденит в сочетании с гепатомегалией позволяет исключить паротитную инфекцию [12, 13]. Причиной ошибки диагностики лейкоза в приведенном наблюдении является отсутствие анализа имевшейся у пациента клинической симптоматики на предмет несоответствий имеющихся у больного симптомов предполагаемому диагнозу.
Клиническое наблюдение № 2
Больная С., 8 месяцев. Направлена участковым врачом на стационарное лечение с диагнозом: «Кишечная инфекция неуточненная, энтерит средней степени тяжести».
Из анамнеза известно, что пациентка заболела 3.06.2021 г. Начало заболевания постепенное. Снизился аппетит, появилось беспокойство, температура тела повысилась до 37,8 °С. Частота стула увеличилась до 5 раз в сутки. Характер стула водянистый, без видимых патологических примесей. Лечилась амбулаторно. Направлена участковым врачом на стационарное лечение в связи с учащением стула и сохранением лихорадки.
При осмотре 10.06.2021 г. в приемном отделении инфекционного стационара состояние – средней тяжести. Температура тела – 37,4 °С. Телосложение правильное, подкожно-жировой слой развит удовлетворительно. Симптомов обезвоживания нет. Кожные покровы бледные. На груди и животе – необильная геморрагическая сыпь до 0,2-0,3 см в диаметре. Увеличение лимфатических узлов шейной группы, подмышечных и паховых до 0,3-0,5 см в диаметре. ЧДД – 34 в минуту. Дыхание пуэрильное, проводится по всем полям, хрипов нет. ЧСС – 132 удара в минуту, тоны сердца приглушены, слабый систолический шум на верхушке и в точке Боткина. Живот умеренно вздут из-за газов, безболезненный при пальпации. Печень выступает на 3 см, селезенка – на 2 см ниже края реберной дуги. Стул кашицеобразный с прожилками крови. Госпитализирована с диагнозом: «Кишечная инфекция неуточненной этиологии». Этот диагноз у врача приемного отделения сомнений не вызывал.
При осмотре в отделении 10.06.2021 г. было обращено внимание на постепенное начало и продолжительность болезни, наличие геморрагического синдрома и гепатоспленомегалии, что не характерно для энтеритов, вызываемых возбудителями кишечной группы.
В общем анализе крови от 10.06.2021 г. количество эритроцитов снижено до 2,1 × 1012/л, гемоглобина – до 60 г/л, тромбоцитов – до 160 × 109/л, количество лейкоцитов увеличено до 29 × 109/л, лимфоцитов – до 89%, обнаружены бластные клетки – 8%.
Больная переведена в гематологическое отделение с диагнозом «ОЛ, период развернутых проявлений». Случай заболевания у больной С. не представлял затруднений для диагностики лейкоза, так как имелся весь симптомокомплекс, характерный для этого заболевания (лимфаденопатия, геморрагический синдром, гепатоспленомегалия). Вовлечение кишечника в патологический процесс при лейкозе не является редкостью и может обуславливаться синдромом энтеропатии вследствие экстрамедуллярного поражения с кровоизлияниями в слизистую оболочку кишечника, активацией условно-патогенных бактерий, входящих в состав микрофлоры кишечника или поступивших в него алиментарным путем [1, 6].
Ошибочная постановка диагноза в стационаре стала очевидной практически сразу. Вероятно, основной причиной ошибки диагностики лейкоза у этой больной является недостаточное знание клиники лейкоза и отсутствие настороженности по отношению к этой патологии у врачей поликлинического звена и непрофильных стационаров, куда пациенты могут поступать с различными диагнозами, учитывая полиморфизм симптомов как в предлейкозном, так и остром периодах болезни.
Заключение
Врачу любой специальности необходимо знать клинику лейкоза. Правильная оценка ранних признаков ОЛ помогает своевременно поставить или заподозрить это заболевание. Учитывая полиморфизм клинической симптоматики ОЛ, больным с анемией, лимфаденопатией, геморрагическим синдромом, гепатоспленомегалией необходимо назначать общий анализ крови с подсчетом тромбоцитов. При ОЛ снижается количество эритроцитов, гемоглобина, нейтрофилов и тромбоцитов, обнаруживаются бластные клетки, увеличивается СОЭ.
Во всех сомнительных случаях ребенок должен быть осмот-рен гематологом.
КОНФЛИКТ ИНТЕРЕСОВ. Авторы статьи подтвердили отсутствие конфликта интересов, о котором необходимо сообщить.
CONFLICT OF INTERESTS. Not declared.
Литература/References
- Маткевич Г. Л., Маякова С. Я. Лейкозы у детей. М.: Практическая медицина, 2009. 384 с. [Matkevich G. L., Mayakova S. A. Leukemia in children. M.: Prakticheskaya medicina, 2009. 384 р. (In Russ.)]
- Детская онкология / Под ред. М. Д. Алиева, В. Г. Полянова, Г. Л. Ментковича, С. А. Маяковой. М.: Издание группы РОНЦ, 2012. 684 с. [Pediatric oncology / Pod red. M. D. Aliyev, V. G. Polyanov, G. L. Mentkovich, S. A. Mayakova. M.: Izdanie gruppy RONC, 2012. 684 р. (In Russ.)]
- Теплякова Е. Д., Сависько А. А., Асланян А. А. Заболеваемость острыми лейкозами у детей в Ростовской области за период 1991-2010 гг. // Фундаментальные исследования. 2012: 2 (2): 363-367. [Teplyakova E. D., Savisko A. A., Aslanyan A. A. Incidence of acute leukemia in children in the Rostov region for the period 1991-2010 // Fundamental’nye issledovaniya. 2012: 2 (2): 363-367. (In Russ.)]
- Hunger S. P., Mullighan C. G. Acute lymphoblastic leukemia in children // N. Engl J. Med. 2015; 373 (16): 1541-1552.
- Казначеев К. С. Сложные вопросы ранней диагностики острого лейкоза у детей // Вестник Новосибирского гос. мед. университета. Серия: Биология. Клиническая медицина. 2011; 9 (2): 211-214. [Kaznacheev K. S. Complex issues of early diagnosis of acute leukemia in children // Vestnik Novosibirskogo gos. med. universiteta. Seriya: Biologiya. Klinicheskaya medicina. 2011; 9 (2): 211-214. (In Russ.)]
- Баровеная Ю. А. Современные аспекты диагностики и лечения острого миелоидного лейкоза у детей (обзор литературы) // Вопросы гематологии/онкологии и иммунологии в педиатрии. 2015; 14 (3): 48-54. [Barovenaya Yu. A. Modern aspects of diagnosis and treatment of acute myeloid leukemia in children (literature review) // Voprosy gematologii/onkologii i immunologii v pediatrii. 2015; 14 (3): 48-54. (In Russ.)].
- Cooper S. L., Brown P. A. Treatment of pediatric acute lymphoblastic leukemia // Pediatr Clin North Am. 2015; 62 (1): 61-73.
- Мамаев Н. Н., Рябов С. И. Гематология. Руководство для врачей. СПб. СпецЛит, 2008. 543 с. [Mamaev N. N., Ryabov S. I. Hematology. Rukovodstvo dlya vrachej. SPb: SpecLit, 2008. 543 р. (In Russ.)]
- Гематология иммунология детского возраста / Под ред. И. А. Алексеева. СПб: Гиппократ, 2009. 1044 с. [Hematology immunology of children / Pod red. I. A. Alekseeva. SPb: Gippokrat, 2009. 1044 р. (In Russ.)]
- Луговая С. А., Почтарь М. Е. Гематологический атлас. 4-е изд., доп. М.: ООО Издательство «Триада», 2016. 434. [Lugovaya S. A., Pochtar M. E. Hematological Atlas 4th edition additional. M.: OOO Izdatel’stvo «Triada», 2016. 434 р. (In Russ.)]
- Hunger S. P., Baruchel A., Biondi A., et al. The thirteenth international childhood acute lymphoblastic leukemia workshop report // Pediatr Blood Cancer. 2013; 60 (2): 344-348.
- Учайкин В. Ф. Руководство по инфекционным болезням у детей. М.: ГЭОТАР-МЕД, 2002. 824 с. [Uchaykin V. F. Guide to infectious diseases in children. M.: GEOTAR-MED, 2002. 824 р. (In Russ.)]
- Харченко Г. А., Кимирилова О. Г. Эпидемический паротит у детей – актуальность проблемы // Детские инфекции. 2017; 16 (3): 28-31. [Kharchenko G. A., Kimirilova O. G. Epidemic mumps in children – the relevance of the problem // Detskie infektsii. 2017; 16 (3): 28-31. (In Russ.)]
Г. А. Харченко, ORCID: 0000-0001-7764-0995, Xarchenkoga@mail.ru
О. Г. Кимирилова, ORCID: 0000-0003-4066-2431, 0lgakim@mail.ru
Федеральное государственное бюджетное образовательное учреждение высшего образования Астраханский государственный медицинский университет Министерства здравоохранения Российской Федерации; 414000, Россия, Астрахань, ул. Бакинская, 121
Сведения об авторах:
Харченко Геннадий Андреевич, д.м.н., профессор, заведующий кафедрой детских инфекций Федерального государственного бюджетного образовательного учреждения высшего образования Астраханский государственный медицинский университет Министерства здравоохранения Российской Федерации; 414000, Россия, Астрахань, ул. Бакинская, 121; Xarchenkoga@mail.ru
Кимирилова Ольга Геннадьевна, к.м.н., доцент кафедры детских инфекций Федерального государственного бюджетного образовательного учреждения высшего образования Астраханский государственный медицинский университет Министерства здравоохранения Российской Федерации; 414000, Россия, Астрахань, ул. Бакинская, 121; 0lgakim@mail.ru
Information about the authors:
Gennady A. Kharchenko, Dr. of Sci. (Med.), Professor, Head of the Department of Pediatric Infections at the Federal State Budgetary Educational Institution of Higher Education Astrakhan State Medical University of the Ministry of Health of the Russian Federation; 121 Bakinskaya str., Astrakhan, 414000, Russia; Xarchenkoga@mail.ru
Olga G. Kimirilova, MD, Associate Professor of the Department of Pediatric Infections at the Federal State Budgetary Educational Institution of Higher Education Astrakhan State Medical University of the Ministry of Health of the Russian Federation; 121 Bakinskaya str., Astrakhan, 414000, Russia; 0lgakim@mail.ru
Ошибки диагностики острого лейкоза у детей грудного возраста (клинические наблюдения)/ Г. А. Харченко, О. Г. Кимирилова
Для цитирования: Харченко Г. А., Кимирилова О. Г. Ошибки диагностики острого лейкоза у детей грудного возраста (клинические наблюдения) // Лечащий Врач. 2023; 1 (26): 7-10. DOI: 10.51793/OS.2023.26.1.001
Теги: детский возраст, лихорадка, слабость, анемия, лейкоз
Лейкоз: причины появления, симптомы, диагностика и способы лечения.
Определение
Лейкозы – большая группа заболеваний кроветворной системы. Кроветворение (гемопоэз) – это многоэтапный процесс образования форменных элементов крови в специализированных органах кроветворения. К форменным элементам крови относятся лейкоциты (белые кровяные тельца), которые участвуют во врожденном и приобретенном иммунитете, эритроциты (красные кровяные тельца), которые осуществляют транспорт кислорода и углекислого газа, и тромбоциты, обеспечивающие свертываемость крови.
 У всех клеток крови есть одна общая «родоначальница» – полипотентная стволовая клетка. Термин «полипотентная» означает, что такая клетка может развиваться в различные виды зрелых клеток.
У всех клеток крови есть одна общая «родоначальница» – полипотентная стволовая клетка. Термин «полипотентная» означает, что такая клетка может развиваться в различные виды зрелых клеток.
Главный орган, где располагаются полипотентные стволовые клетки, — костный мозг, который как раз и поражается при лейкозах.
В результате повреждения (мутации) в генетическом материале полипотентной стволовой клетки-предшественницы нарушается процесс созревания клеток. При остром лейкозе возникает большое количество незрелых (бластных) клеток, из которых образуется опухоль, разрастается в костном мозге, замещает нормальные ростки кроветворения и имеет тенденцию к метастазированию – распространению с током крови или лимфы в здоровые органы. При хронических лейкозах заболевание течет годами, происходит частичная задержка созревания клеток и опухоль образуется из созревающих и зрелых клеток.
 Причины возникновения лейкозов
Причины возникновения лейкозов
К факторам риска развития лейкоза относятся:
- ионизирующая радиация: лучевая терапия по поводу других опухолей, облучение на рабочем месте, ультрафиолетовое излучение;
- воздействие на организм химических канцерогенных веществ;
- некоторые вирусы: HTLV (Т-лимфотропный вирус человека);
- бытовые факторы: добавки в пищевые продукты, курение, плохая экологическая обстановка;
- наследственная предрасположенность к раковым заболеваниям.
Классификация лейкозов
Лейкозы подразделяют на острые и хронические, а по типу поражения ростка кроветворения — на лимфоидные и миелоидные. Острый лейкоз никогда не переходит в хронический, а хронический не может стать острым (но его развитие может перейти в стадию бластного криза).
В зависимости от преобладания тех или иных опухолевых клеток выделяют бластные, цитарные и недифференцированные лейкозы.
Острый лимфобластный лейкоз (ОЛЛ) диагностируется у лиц любого возраста, начиная с младенческого и заканчивая пожилым, однако пик заболеваемости приходятся на детский возраст (60% пациентов с ОЛЛ моложе 20 лет). Острый лимфобластный лейкоз – самая распространенная опухоль кроветворной ткани у детей, которая составляет 30% всех злокачественных опухолей детского возраста. Заболеваемость ОЛЛ у пациентов пожилого возраста неуклонно возрастает: так, ежегодная частота ОЛЛ увеличивается с 0,39 случая на 100 тыс. населения в возрасте 35–39 лет, до 2,1 случая на 100 тыс. населения в возрасте ≥85 лет. Кроме того, приблизительно 30% ОЛЛ диагностируются в возрасте ≥60 лет.
Острым миелоидным лейкозом (ОМЛ) страдает в среднем 3-5 человек на 100 тыс. населения в год. Заболеваемость резко возрастает в возрасте старше 60 лет и составляет 12–13 случаев на 100 тыс. населения у лиц в возрасте старше 80 лет.
Хронический лимфобластный лейкоз (ХЛЛ) – самый частый вид лейкозов у взрослых, в то время как у детей этот вид опухоли не регистрируется. В европейских странах его частота составляет 4 случая на 100 тыс. населения в год и непосредственно связана с возрастом. У лиц старше 80 лет она составляет более 30 случаев на 100 тыс. в год.
Хронический миелолейкоз (ХМЛ) – редкое заболевание: 0,7 на 100 тыс. взрослого населения, пик заболеваемости приходится на 50-59 лет, однако до 33% больных ОМЛ — люди моложе 40 лет.
Симптомы лейкозов
Острый лейкоз в большинстве случаев дебютирует резко — внезапно повышается температура, появляются озноб, боль в горле, в суставах, отмечается резко выраженная слабость. Реже острый лейкоз может проявиться кровотечением. Иногда острый лейкоз начинается с постепенного ухудшения состояния больного, появления невыраженной боли в суставах и костях, кровоточивости. В единичных случаях возможно бессимптомное начало заболевания. У многих больных увеличиваются лимфоузлы и селезенка.
При хроническом лейкозе на начальной стадии, которая длится от года до трех лет, пациенты могут ни на что не жаловаться. Иногда беспокоят слабость, потливость, частые простудные заболевания, могут отмечаться тупые, ноющие боли в костях.
Симптоматика лейкозов зависит от того, какой росток кроветворения поражен.
При подавлении эритроидного ростка, дающего красные кровяные тельца, возникает анемия и гемическая гипоксия (снижение количества кислорода в крови). При этом пациенты отмечают слабость, утомляемость, бледность кожных покровов.
При поражении мегакариоцитарного ростка падает количество тромбоцитов, поэтому возникает кровоточивость десен, слизистой оболочки носа, пищеварительного тракта, образуются синяки, кровоизлияния в различные органы. При прогрессировании лейкоза могут развиваться массивные кровотечения в результате ДВС синдрома (синдрома диссеминированного внутрисосудистого свертывания).
В результате недостатка лейкоцитов и снижения иммунитета развиваются инфекционные осложнения различной степени тяжести, что чаще всего проявляется лихорадкой.
Могут возникать язвенно-некротическая ангина, перитонзиллярные абсцессы, некротический гингивит, стоматит, пиодермия, параректальные абсцессы, пневмония, пиелонефрит. Существует значимый риск тяжелого течения инфекционных осложнений вплоть до развития сепсиса.
С током крови и лимфы опухолевые клетки попадают в здоровые органы, нарушают их структуру и функцию — наиболее подвержены метастатическим процессам печень, селезенка, лимфатические узлы, но метастазы могут поражать и кожу, и мозговые оболочки, и почки, и легкие.
Основные причины летальности у пациентов с лейкозом связаны с тем, что осложнения могут спровоцировать развитие сепсиса, полиорганную недостаточность, кровоизлияния в различные органы. Острый лейкоз без лечения приводит к смертельному исходу в течение нескольких недель или месяцев.
Диагностика лейкозов
Диагностика лейкозов основывается на оценке морфологических особенностей клеток костного мозга и периферической крови. Поэтому всем пациентам с подозрением на лейкоз назначают общий анализ крови с подсчетом лейкоцитарной формулы и определением числа тромбоцитов.
Для определения объема опухолевого поражения, вероятности развития синдрома лизиса опухоли рекомендовано выполнение общетерапевтического биохимического анализа крови: АСТ, АЛТ, общий билирубин, глюкоза, мочевина, креатинин, общий белок, ЛДГ, магний, натрий, калий, кальций.
Билирубин общий (Bilirubin total)
Синонимы: Общий билирубин крови; Общий билирубин сыворотки.
Totalbilirubin; TBIL.
Краткая характеристика определяемого вещества Билирубин общий
Билирубин – пигмент �…
Глюкоза (в крови) (Glucose)
Материал для исследования
Сыворотка или плазма крови. Если нет возможности центрифугировать пробу через 30 минут после взятия для отделения сыворотки/плазм…
Мочевина (в крови) (Urea)
Синонимы: Диамид угольной кислоты; Карбамид; Мочевина в крови; Азот мочевины.
Urea nitrogen; Urea; Blood Urea Nitrogen (BUN); Urea; Plasma Urea.
Краткая характеристика аналита Мочевина
Моче�…
Креатинин (в крови) (Creatinine)
Синонимы: Анализ крови на креатинин; Сывороточный креатинин; Креатинин сыворотки, оценка СКФ. Сreat; Сre; Blood Creatinine; Serum Creatinine; Serum Creat.
Краткая характеристика определя�…
Общий белок (в крови) (Protein total)
Синонимы: Общий белок сыворотки крови; Общий сывороточный белок.
Total Protein; Serum Тotal Protein; Total Serum Protein; TProt; ТР.
Краткая характеристика определяемого вещества Общий бе�…
Магний в сыворотке (Magnesium, Serum; Мg)
Синонимы: Анализ крови на магний; Общий магний; Ионы магния. Serum Magnesium Test; Magnesium test; Magnesium, blood; Serum magnesium.
Краткая характеристика определяемого аналита Магний
…
Калий, натрий, хлор в сыворотке крови (К+, Potassium, Na+, Sodium, Сl-, Chloride, Serum)
Синонимы: Анализ крови на электролиты; Электролиты в сыворотке крови. Electrolyte Panel; Serum electrolyte test; Sodium, Potassium, Chloride; Na/K/Cl.
Краткая характеристика определяемых �…
Кальций общий (Ca, Calcium total)
Общий кальций – основной компонент костной ткани и важнейший биогенный элемент, обладающий важными структурными, метаболическими и регуляторными функциями в …
Для определения вероятности развития тяжелых коагуляционных нарушений (как геморрагических, так и тромботических) рекомендовано исследование свертывающей системы крови.
Гемостазиограмма (коагулограмма), скрининг
Синонимы: Гемостазиограмма, коагулограмма.
Coagulation studies (coagulation profile, coag panel, coagulogram).
Состав профиля:
№ 2 Протромбин (протромбиновое время, протромбин (по Квику), МНО…
С целью исключения поражения почек выполняют общий анализ мочи.
Диагностику хронического лейкоза проводят с помощью иммунофенотипического исследования лимфоцитов крови (ИФТ) методом проточной цитометрии.
Рекомендовано проведение стернальной пункции (пункции грудины) для получения цитологического препарата костного мозга и цитологическое и цитохимическое исследование мазка с целью уточнения диагноза и определения прогноза.
В ряде случаев показана биопсия опухолевого образования или лимфатического узла (или другого метастатического очага) и патологоанатомическое исследование полученного биопсийного материала.
Кроме того, врач может рекомендовать проведение иммунофенотипирования гемопоэтических клеток-предшественниц в костном мозге, цитогенетическое исследование аспирата костного мозга, молекулярно-генетические исследования мутаций в генах и др.
Из инструментальных методов диагностики проводятся:
- эхокардиография для оценки функционального состояния сердечной мышцы;
Эхокардиография
Исследование, позволяющее оценить функциональные и органические изменения сердца, его сократимость, а также состояние клапанного аппарата.
КТ головного мозга и черепа
Сканирование головного мозга, черепа и окружающих их тканей, позволяющее диагностировать различные патологии.
К каким врачам обращаться
Лечением острых и хронических лейкозов занимаются
врачи-онкологи
и онкогематологи. По показаниям пациентам могут потребоваться консультации смежных специалистов:
врача-невролога
, офтальмолога, оториноларинолога, всем женщинам рекомендована консультация врача —
акушера-гинеколога
.
Лечение лейкозов
Лечение лейкозов проводится в стационаре. Медикаментозное воздействие на опухоль специальными препаратами, губительно действующими на быстро делящиеся клетки, называется химиотерапией. При лечении острых лейкозов химиотерапию проводят в несколько этапов: индукция ремиссии, консолидация (закрепление) ремиссии, поддерживающая терапия и профилактика нейролейкемии (метастазирования опухолевых лейкозных клеток в головной и спинной мозг).
Период индукции ремиссии — это начальный этап, цель которого максимально уменьшить опухолевую массу и достичь ремиссии. Обычно для этого требуется 1-2 курса химиотерапии. Далее идет консолидация достигнутого эффекта — наиболее агрессивный и высокодозный этап лечения, задачей которого является по возможности еще большее уменьшение числа остающихся после индукции лейкемических клеток. Этот этап также занимает 1-2 курса. За ним следует противорецидивное или поддерживающее лечение. При некоторых вариантах острых лейкозов требуется профилактика или, при необходимости, лечение нейролейкемии.
Для разных видов лейкозов у разных возрастных групп профессиональными сообществами разработаны схемы химиотерапии.
При лечении ХМЛ основными препаратами выбора являются ингибиторы тирозинкиназы, применяемые в непрерывном режиме – ежедневно, длительно, постоянно. Перерывы в приеме могут способствовать снижению эффективности терапии и прогрессированию заболевания. В случае неэффективности терапии может быть проведена трансплантацию гемопоэтических стволовых клеток или костного мозга.
В терапии ХЛЛ цели и схемы терапии химиотерапии определены возрастом пациента, числом и тяжестью сопутствующих заболеваний. Разработаны протоколы лечения для разных пациентов, в том числе схемы моно- и полихимиотерапии. Для профилактики инфекционных осложнений применяют внутривенное введение иммуноглобулина, рекомендована вакцинация от гриппа и пневмококковой инфекции.
Помимо химиотерапии пациенту может потребоваться трансфузионная терапия: переливание эритроцитарной массы, тромбоцитарной массы, изотонических растворов.
При присоединении инфекций показана антибиотикотерапия. На фоне лечения могут возникать тромботические осложнения, что требует антикоагулянтной терапии. Пациентам высокого риска в связи с вероятностью рецидива лейкоза применяют трансплантацию гемопоэтических стволовых клеток.
Осложнения
Основные осложнения лейкозов — инфекционные, вплоть до сепсиса, и тромботические и/или геморрагические осложнения с развитием синдрома диссеминированного внутрисосудистого свертывания. При метастазировании опухолевых клеток развивается полиорганная недостаточность, в т.ч. лейкозные клетки могут попадать в оболочки головного и спинного мозга, инфильтрировать периферические нервы с развитием разнообразных двигательных и чувствительных нарушений (лейкемоидная инфильтрация). Кроме того, встречается так называемый «синдром лизиса опухоли» — сложный процесс, возникающий в результате спонтанного или вызванного лечением разрушения большого числа опухолевых клеток с выходом внутриклеточного содержимого в кровоток. При этом может развиться острая почечная недостаточность. Летальность при лизисе опухоли достигает 70%.
Профилактика лейкозов
Так как причины возникновения лейкозов не установлены, методов специфической профилактики до сих пор не существует.
Однако доказано, что между курением и риском развития острого лейкоза существует дозовая зависимость, которая особенно очевидна для лиц старше 60 лет.
Ряд исследователей предполагают, что около 20% случаев ОМЛ являются следствием курения.
Источники:
- Клинические рекомендации «Острые миелоидные лейкозы». Общероссийский национальный союз «Ассоциация онкологов России», некоммерческое партнерство содействия развитию гематологии и трансплантологии костного мозга «Национальное гематологическое общество», 2020.
- Клинические рекомендации «Острые лимфобластные лейкозы». Общероссийский национальный союз «Ассоциация онкологов России», некоммерческое партнерство содействия развитию гематологии и трансплантологии костного мозга «Национальное гематологическое общество», 2020.
- Клинические рекомендации «Хронический лимфоцитарный лейкоз/ лимфома из малых лимфоцитов». Общероссийский национальный союз «Ассоциация онкологов России», некоммерческое партнерство содействия развитию гематологии и трансплантологии костного мозга «Национальное гематологическое общество», региональная общественная организация «Общество онкогематологов», 2020.
- Клинические рекомендации «Хронический миелолейкоз у взрослых». Национальное гематологическое общество, 2017.
ВАЖНО!
Информацию из данного раздела нельзя использовать для самодиагностики и самолечения. В случае боли или иного обострения заболевания диагностические исследования должен назначать только лечащий врач. Для постановки диагноза и правильного назначения лечения следует обращаться к Вашему лечащему врачу.
Для корректной оценки результатов ваших анализов в динамике предпочтительно делать исследования в одной и той же лаборатории, так как в разных лабораториях для выполнения одноименных анализов могут применяться разные методы исследования и единицы измерения.
Информация проверена экспертом

Лишова Екатерина Александровна
Высшее медицинское образование, опыт работы — 19 лет
Поделитесь этой статьей сейчас
Рекомендации
-
3737
09 Февраля
-
3735
09 Февраля
-
3756
08 Февраля
Похожие статьи
Рак кишечника
Рак кишечника: причины появления, симптомы, диагностика и способы лечения.
Лейкоз – характеристика заболевания у детей
У детей лейкемия подразумевает под собой заболевание крови, которое является злокачественным. Во время острого лейкоза у детей клетки формируются неправильно. Клетка, которая только зарождается, не развивается надлежащим образом, она является бластом. Вместо лейкоцитов, которые являются здоровыми, появляются атипичные патогены. Это происходит во время процесса деления. Слой бласта со временем вытесняет тот ряд лейкоцитов, который является нормальным.
Поскольку вместо здоровых клеток появляются аномальные, то возникает лейкоз крови у детей. Опухоль размещается в каком-то одном участке и там происходит ее развитие, во время которого размеры новообразования увеличиваются. Заболевание приобретает злокачественный характер. Процесс очень быстро распространяется и затрагивает другие органы. Довольно быстро у детей появляются метастазы в тканях. Чтобы не дать раковым клеткам дальше развиваться, нужно принимать определенные лекарства, использовать специальные методики. Аномальный процесс не может прекратиться сам.
Лимфобластный лейкоз у детей особо агрессивен. Здесь отмечают быстрый темп распространения метастазов, при этом страдают ткани, появляются очаги вторичного типа, патологические процессы. Если начинать лечить лейкоз, когда он только начинает развиваться, то можно говорить о положительном результате. Если срок более поздний, то здесь уже сложнее вылечить заболевание. Процесс будет длиться дольше. Ребенку проще и легче даются разные процедуры, он слушает специалиста и соблюдает все указания. Это помогает добиться выздоровления.
Чаще всего диагностируют острый лейкоз у детей возрастом 2-5 лет. Но практика показывает, что иногда болезнь может возникать у подростков, грудничков. Обычно диагностируют лейкоз острой формы, когда симптомы проявляются очень стремительно. Педиатрия сегодня пытается сделать все возможное, чтобы предотвратить заболеваемость детей.
Причины развития болезни у детей
У детей болезнь может возникать, если в организме мутируют клетки, относящиеся к лейкоцитарному ряду. Кроме того, следует учитывать такие причины острого лейкоза у детей, способные спровоцировать патологический процесс:
- предрасположенность из-за наследственности;
- радиационное излучение;
- вирус, который может спровоцировать лейкемию;
- лекарства специфического характера;
- нарушение, которое происходит в хромосомах. Эта причина лейкоза у детей считается главной;
- применение химиотерапии, лучевой терапии (их могут использовать, когда лечат онкологию другого органа);
- отсутствие в пище клетчатки растительного вида;
- канцерогены, токсические вещества;
- постоянное нахождение ребенка в местности, которая отличается плохой экологией.
Заболевание может возникнуть в результате воздействия любой из этих причин рака крови у детей или нескольких.
Классификация болезни
Классификация может создаваться в зависимости от структурных характеристик, состава раковых клеток, времени протекания болезни. Если говорить о патологическом процессе и его скорости развития, то здесь стоит выделить два типа – острый (весь процесс длится около 2 года), а также хронический (болезнь развивается больше 2 года). Первый вид опухоли разделяют на острый лимфобластный лейкоз у детей и нелимфобластный тип. При первом варианте лимфоциты аномального типа делятся очень стремительно.
Еще ученые разделяют лейкоз на две формы:
- лимфоцитарный, при котором лимфоциты размножаются с особой активностью;
- миеловидный, при котором активно делятся гранулоциты.
Есть еще бифенотипический лейкоз острой формы, его главное отличие – наличие маркеров.
Если изучить более подробно болезнь, ее клиническую картину, то здесь выделяют определенные этапы:
- острая фаза. Здесь появляются признаки онкологии до того, как после терапии начинается улучшение;
- ремиссия лечения, она может быть полной либо частичной;
- рецидив.
Признаки болезни у детей
У детей лейкоз развивается очень быстро. Первый признак лейкоза у детей – это то, что гематогенная структура крови нарушается. Это уже видно на том этапе, когда мутируют клетки. Но на первый взгляд нельзя увидеть признак лейкоза. Для этого нужно сделать лабораторную диагностику.
Но в детском возрасте симптомы лейкоза крови у детей могут напоминать другие болезни:
- ребенок вялый. После того как была осуществлена физическая нагрузка, отмечается быстрая усталость;
- появляется бессонница, малыш не может спать по тому же режиму, как раньше;
- нет аппетита либо он пропадает частично;
- возникают болевые ощущения в области суставов, костей;
- температура достигает термальных показателей.
Появляются признаки лейкоза у детей, которые напоминают геморрагический, интоксикационный синдром:
- кожа и слизистая становятся желтоватыми, бледными;
- появляются заболевания инфекционного характера внутри ротовой полости (стоматит, тонзиллит, гингивит);
- увеличиваются слюнные железы, селезенка, печень, лимфоузлы;
- на коже может появляться сыпь;
- в верхних слоях дермы происходит кровоизлияние капилляров;
- из носа идет кровь;
- кровоизлияние происходит также внутри организма, в органах;
- отмечается анемия крови в хронической форме;
- появляются симптомы острого лейкоза у детей, указывающие на то, что ритм сердца нарушен;
- состояние лихорадки;
- выделяется из организма больше пота;
- ребенок теряет вес;
- его часто тошнит и он хочет вырвать;
- наблюдается задержка развития, это касается умственных показателей, а также физического состояния;
- воспалительный процесс внутри организма;
- наличие головных болей, они достаточно сильные. Еще у ребенка может кружиться голова;
- его нервные окончания, мышцы не настолько чувствительны.
Когда следует обратиться к врачу
К специалисту стоит обращаться незамедлительно, как только у ребенка появились симптомы лейкоза у детей. Если ребенок чувствует себя не очень хорошо, то лучше сдать анализы, обследоваться, чтобы исключить такой диагноз, как лейкоз.
Диагностика заболевания
Распознать заболевание на ранних стадиях можно, но для этого нужно провести ребенку расширенное обследование, то есть необходимо изучить весь организм. Ведь изначально отсутствуют какие-либо симптомы, поэтому очень сложно определить лейкоз у ребенка. Первым специалистом, который занимается диагностикой лейкоза у детей, является педиатр. Если врач сомневается в диагнозе, он сразу же направляет пациента к онкологу, для его подтверждения.
Что касается диагностики, то здесь выполняется ряд процедур (инструментальные, лабораторные):
- общий анализ крови. Он дает возможность определить лейкоцитоз, анемию, ретикулоцитопению, отклонения, имеющие отношение к СОЭ;
- миелограмма, пункция костного мозга. Благодаря таким процедурам можно понять, на каком уровне находятся бластные клетки. Если их показатель равняется 30% и больше, тогда врач может говорить о лейкозе у детей;
- трепанобиопсия. Она позволяет получить более точные данные;
- иммунологические, цитогенетические, цитохимические обследования;
- УЗИ брюшной полости, всех органов, также изучается с помощью такого исследования малой таз. УЗИ дает понять, есть ли в этих органах нарушения;
- КТ показывает, есть ли в организме метастазы;
- рентгенография черепа. С ее помощью врач видит, была ли нарушена структура головного мозга, его тканей;
- обследование рака крови у детей у более узких врачей. Речь идет об офтальмологе, неврологе.
Лечение детского лейкоза
Если миелобластный лейкоз у детей находится на ранних стадиях, то его можно вылечить, но после лейкоза обычно человеку присваивают инвалидность. Пациенты должны пребывать в специализированном онкологическом центре. Ребенок должен находиться в условиях, которые отличаются полной стерильностью. Этого достичь в домашних условиях нереально. Поэтому больного помещают в специальный бокс, где и осуществляют лечение острого лейкоза у детей. Что касается протокола лечения, то здесь нужен индивидуальный подход, ведь каждый случай отличается от других. Поэтому лекарственная доза должна обсуждаться непосредственно на приеме у врача. Во время лечения рацион питания ребенка должен быть полноценным и сбалансированным.
Чтобы вылечить рак крови у детей, используют полихимиотреапию. Данный метод способен блокировать раковые клетки, не давать им разрастаться. Для этого применяют специальные препараты, которые относятся к группе цитостатиков. Доза здесь подбирается отдельно в каждом случае, все зависит от характера болезни, стадии. Изначально специалист должен довести заболевание до стадии ремиссии. После чего нужно закрепить этот период. А затем пациенту назначают специальную терапию, которая способна поддержать маленького пациента. Здесь также применяются профилактические меры, чтобы предотвратить рецидив лейкемии. Если после того как было осуществлено лечение лейкоза у детей, появились осложнения, то назначают терапию, которая позволит купировать симптоматику.
Еще в дополнение назначают профилактику. То есть, ребенку делают прививки от оспы, БЦЖ, вводят интерферон, клетки лейкоза, имеющие лимфоциты. Также врач прописывает курс терапии, чтобы убрать анемию, нарушения, происходящие в крови, ее структуре. Также применяются антибактериальные лекарства, чтобы купировать инфекционный процесс внутри организма. Могут переливать кровь, лимфу. Во избежание рецидива заболевания за ребенком постоянно наблюдают. После того как лечение острого миелобластного лейкоза у детей закончится, маленький пациент регулярно должен посещать специалистов клиники.
Прогноз выживаемости
Прогноз при остром лейкозе у детей будет напрямую зависеть от возраста ребенка, на какой стадии находится болезнь, насколько сильный организм (его физические показатели), как себя чувствует пациент. Прогноз будет неблагоприятным в следующих ситуациях:
- малышу еще нет 2 года либо школьнику уже исполнилось 10 лет;
- отмечается лимфаденопатия, а также наличие гепатоспленомегалии;
- во время лечения присутствует нейролейкоз;
- диагностируется такой тип болезни, как B-, T-клеточный.
После прохождения лечения у пациентов, которые пребывают в возрастной группе 2-10 лет, прогноз при лейкозе у детей будет положительным. Но это касается тех случаев, когда патологию определили на ранних стадиях. Что касается пола, то шансы выздороветь немного выше у девочек. В случае отсутствия надлежащего лечения у детей наступает летальный исход. Поэтому очень важно вовремя обратиться к специалисту, а не использовать дома народные методы, осуществлять самостоятельное лечение.
Если на протяжении 7 лет не возникают рецидивы детского лейкоза, то это может говорить о том, что ребенок полностью выздоровел. Однако нельзя ни в коем случае останавливать наблюдение за его состоянием. Что касается вакцинации, то ее профилактика должна составляться, учитывая историю болезни пациента.
Как записаться к онкологу
Если вдруг вы заметили симптомы у детей лимфобластного лейкоза, тогда лучше обратиться сразу к специалисту, чтобы исключить диагноз либо подтвердить его. Онкологи, работающие в АО «Медицина» (клиника академика Ройтберга) назначат все необходимые исследования, чтобы убедиться в диагнозе. После чего с учетом клинической картины заболевания, ее особенностей, стадии развития назначает эффективное лечение. Следует помнить, что прогноз жизни при остром лейкозе у детей будет положительным только в том случае, когда за помощью обращаются своевременно.
Записаться к врачам можно несколькими способами – приехать в клинику, которая находится рядом со станциями метро Маяковская, Чеховская, Новослободская, Белорусская, заполнить специальную форму на сайте или по телефону +7 (495) 775-73-60. В телефонном режиме вас могут записать на прием, проконсультировать и предоставить всю необходимую информацию. Лейкоз у детей лечится. Главное, вовремя обратиться за помощью.
Лицензии и сертификаты
- Case Report
- Open Access
- Published: 15 October 2009
- Farina Arreguin-Gonzalez1,
- Carlos A Rodriguez-Osorio2,
- Stanislaw Sadowinski3,
- Rosana Pelayo4 &
- …
- Aurora Medina-Sanson1
Cases Journal
volume 2, Article number: 154 (2009)
Cite this article
-
7171 Accesses
-
9 Citations
-
Metrics details
Abstract
Acute leukemia, the most common form of cancer in children, accounts for approximately 30% of all childhood malignancies, with acute lymphoblastic leukemia being five times more frequent than acute myeloid leukemia. Lineage switch is the term that has been used to describe the phenomenon of acute leukemias that meet the standard French-American-British system criteria for a particular lineage (either lymphoid or myeloid) upon initial diagnosis, but meet the criteria for the opposite lineage at relapse. Many reports have documented conversions of acute lymphoblastic leukemia to acute myeloid leukemia.
Here, we report the case of a 4-year-old child with acute myeloid leukemia, which upon relapse switched to acute lymphoblastic leukemia. The morphologic, phenotypic, and molecular features suggest the origin of a new leukemic clone.
Introduction
Leukemia is a group of malignant diseases of the hematopoietic system characterized by the uncontrolled overproduction of either immature (acute leukemia) or terminally differentiated (chronic leukemia) leukocytes.
Acute leukemia, the most common form of cancer in children, accounts for approximately 30% of all childhood malignancies [1]. A frequency of ≥ 20% blasts is required to confirm a diagnosis of acute leukemia. The first comprehensive morphologic classification system for acute leukemias was the French-American-British (FAB) classification, which was established in 1976 and revised in 1985. Other recommended tests are immunophenotyping, cytogenetics, and molecular genetics tests [2].
Acute leukemias can be further sub classified by determining whether the malignant leukocytes are of myeloid origin (cells of granulocyte, monocyte, erythroid, or megakaryocyte lineage) or lymphoid origin (B-cells, T-cells) [3].
Overall survival for patients with acute lymphoblastic leukemia is approximately 80%; currently for acute myeloid leukemia (AML), most large studies show a five-year event-free-survival rate of almost 50%, despite the fact that between one third and one half of patients with AML experience relapse, and no standard therapy is recognized for patients with relapsed and/or refractory AML. Relapses mostly exhibit the same FAB subtype. Patients with relapsed/refractory AML usually respond less well and for a shorter duration to reinduction therapies. This clearly points to the induction of drug-resistant mechanisms and is, in part, related to specific chromosomal abnormalities and the duration of the first remission [4, 5].
However, unexplained variability in clinical course still exists among some individuals within defined risk-group strata.
«Lineage switch» is the term that has been used to describe the phenomenon of acute leukemias that meet standard FAB criteria for a lineage (lymphoid or myeloid) at initial diagnosis but upon relapse meet the criteria for the opposite lineage [6].
Most reports of lineage switching have demonstrated acute lymphoblastic leukemia (ALL) to acute myeloid leukemia (AML) conversions [7–9]. The prognosis for these patients is variable, and there is no standard treatment for them.
Case presentation
A 4-year-old Mexican Mestizo (Hispanic) male was admitted to the Hospital Infantil de México Federico Gómez presenting a 30-day history of fever, pallor, and enlarged cervical lymph nodes.
The initial laboratory findings revealed a white blood cell count of 4,300/mm3, with 16% blasts in the peripheral blood, 8.8 g/dL hemoglobin, and a platelet count of 49,000/mm3.
Cerebrospinal fluid was negative for blast cells. The bone marrow was hypercellular, with 64% blasts, which were large in size and showed low nucleus: cytoplasm ratio, monocytoid features, and blue-gray cytoplasm; > 80% of the blast cells were monoblasts, and the rest were promonocytes or monocytes (Figure 1). The surface immunophenotype was CD45+ (91.1%), CD15+ (95.4%), CD14-/+ (37.7%), CD13+/- (78.5%), HLA-DR+/- (78.5%), CD22-/+ (42.8%), CD10— (2.1%) CD19— (2.5%) CD20— (0.7%) CD7— (0.07%), CD3-/+ (19.4%), and CD33— (7.9%) (Figure 2).
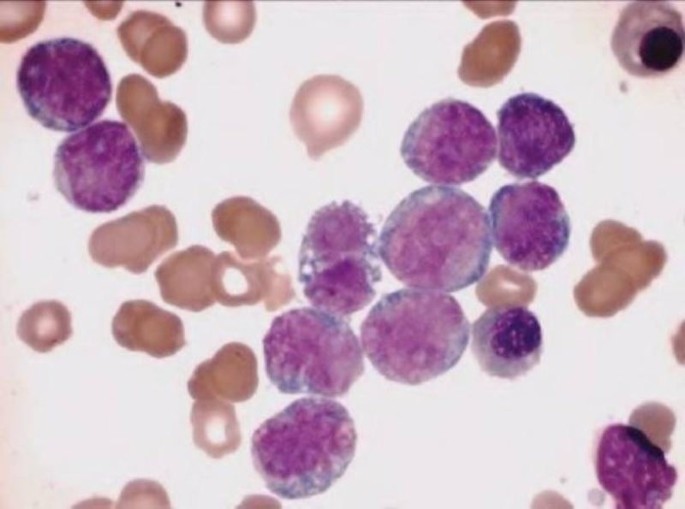
Bone marrow aspiration at AML diagnosis. Blast cells were large in size and had a low nucleus:cytoplasm ratio, monocytoid aspect, and blue-gray cytoplasm. Wright stain, 1000×.
Full size image
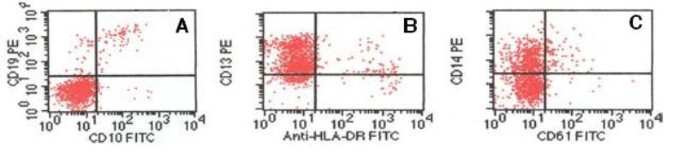
Flow cytometry AML at diagnosis. Immunophenotyping of leukemic cells by flow cytometric analyses revealed myeloid blasts negative for CD10 and CD19 (A) but positive for CD13 (B) and CD14 (C).
Full size image
Cytochemistry was positive for myeloperoxidase (MPO). Conventional cytogenetic analysis using direct and 24-hour unstimulated and unsynchronized cultures revealed a 46 XY karyotype. A diagnosis of M5 AML was made. The patient started chemotherapy with a modified MRC-10 protocol: daunorubicin 50 mg/m2 (days 1, 3 & 5); cytarabine 100 mg/m2 in a 1-hour infusion every 12 hours through days 1 to 10; and etoposide 100 mg/m2 in a 4-hour infusion every 24 hours through days 1 to 5.
Due to oral candidiasis and an episode of sepsis secondary to Acinetobacter iwofii, the patient was treated with meropenem, amphotericin, and fluconazole.
Remission was determined by marrow aspiration with no blast cells at day 27th after first chemotherapy cycle.
Thirty days after the first course of chemotherapy, a second chemotherapy cycle was administered without complications daunorubicin 50 mg/m2 days 1, 3 & 5; cytarabine 100 mg/m2 in a 1-hour infusion every 12 hours through days 1 to 4; and etoposide 100 mg/m2 in a 4-hour infusion every 24 hours through days 1 to 5.
A third cycle: etoposide 100 mg/m2 in a 4-hour infusion every 24 hours on days 1-5 and cytarabine 1000 mg/m2 in a 1-hour infusion every 12 hours (days 1-3) was stopped on day 3 because of a life-threatening episode of pneumonia; the patient was transferred to the ICU and supported by mechanical ventilation. Because a lung CT scan showed features of a right lung abscess, a medium and right lobectomy was performed.
Upon clinical and hematologic recovery, the patient received a fourth cycle of chemotherapy with mitoxantrone 10 mg/m2 in a 1-hour infusion every 24 hours (days 1-5) and cytarabine 1000 mg/m2 in a 1-hour infusion every 12 hours (days 1-3). Two weeks later, an episode of fever, along with neutropenia and mucositis, was recorded, and the patient was treated with cefepime and amikacin.
The patient received a fifth cycle or chemotherapy: etoposide 100 mg/m2 in a 4-hour infusion every 24 hours for 5 days and cytarabine 1000 mg/m2 in a 1-hour infusion every 12 hours for 3 days.
Seven months later, after the diagnosis, the chemotherapy protocol was completed with a clearance of blast cells.
Two months later, the patient presented at the hospital with petechiae. Initial laboratory data revealed a white blood cell count of 7,700/mm3, 13% blast cells, 13.7 g/dL hemoglobin, and a platelet count of 33,000/mm3. The marrow was hypercellular and contained 85% blasts. In contrast to the previous neoplasm, the blasts in the current specimen were smaller, with a predominance of small cells, scanty cytoplasm, moderate cytoplasmic basophilia, and variable cytoplasmic vacuolation (Figure 3), along with the immunophenotype CD45+ (81.6%), CD10+ (77%), CD19+ (89%), CD22— (0%), CD20— (3.7%), CD15— (0%), CD14— (4.5%), CD7— (7.8%), CD3— (13.1%), CD13— (0.27%), CD33— (0.62%), and HLA-DR— (0.2%) (Figure 4). These lymphoblasts were negative for myeloperoxidase (data not shown).
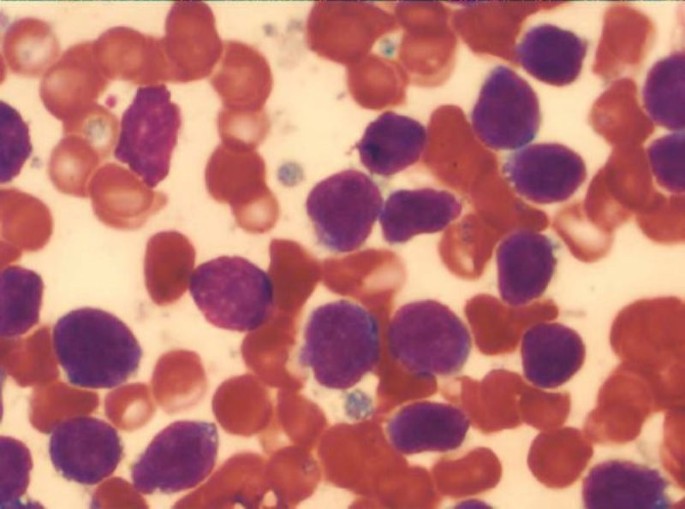
Bone marrow aspiration at relapse. Blasts were smaller, with a predominance of small cells, scanty cytoplasm, moderate cytoplasmic basophilia, and variable cytoplasmic vacuolation. Wright stain, 1000×.
Full size image
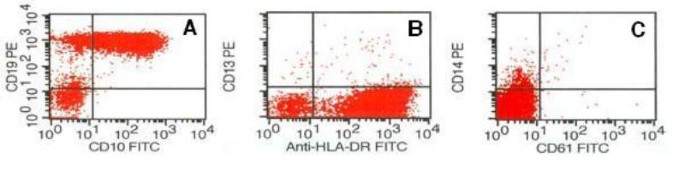
Flow cytometry at relapse. Flow cytometric analyses of leukemic cells at relapse. Blast cells were identified as lymphoblasts because the immunophenotype was positive for CD10 and CD19 (A) but negative for CD13 and CD14 (B, C).
Full size image
The patient was diagnosed with pro-B lymphoblastic leukemia. Cytogenetic studies performed on the marrow aspirate revealed a karyotype of 46 XY.
We tried to induce remission with the national protocol for acute lymphoblastic leukemias, adapted from TOTXV protocol from St Jude Children’s Research Hospital with vincristine 2 mg/m2 once a week for four weeks, daunorubicin 30 mg/m2 once a week for two weeks, L-asparaginase 10,000 IU/m2 three times a week for a total of nine doses, and dexamethasone 6 mg/m2 for four weeks. However, the bone marrow examination on the 14th and 21th days of induction showed persisting infiltration with 50% blasts, and the patient remained pancytopenic. After an episode of fever, abdominal pain, and diarrhea, antibiotic therapy was conducted.
As the bone marrow examination showed persisting infiltration, we indicated cycle 1 and 2 to myeloid leukemia (described above). This scheme of chemotherapy is used in our hospital for patients with early relapse or refractory acute leukemia. After that, the patient had infectious complications, followed by development of tumor lysis syndrome and septic shock. Chemotherapy was stopped and the patient was transferred to the ICU and supported by mechanical ventilation. After clinical recovery, the bone marrow showed persisting infiltration with 85% blasts, and the patient’s parents asked for palliative care; the child was cared for at home for four weeks under the surveillance of a palliative care clinic. Despite supportive measures, he died six months after the diagnosis of relapse. No autopsy was authorized by the family.
In order to investigate if a B lymphoid transcription factor was already expressed at the time of remission of the myeloid leukemia, we performed detection of PAX5 (B-cell-specific activator protein for B cells, including B-lymphoblastic neoplasms) [10], in bone marrow biopsy taken at the end of the first scheme of chemotherapy, when the patient started surveillance (Figure 5).
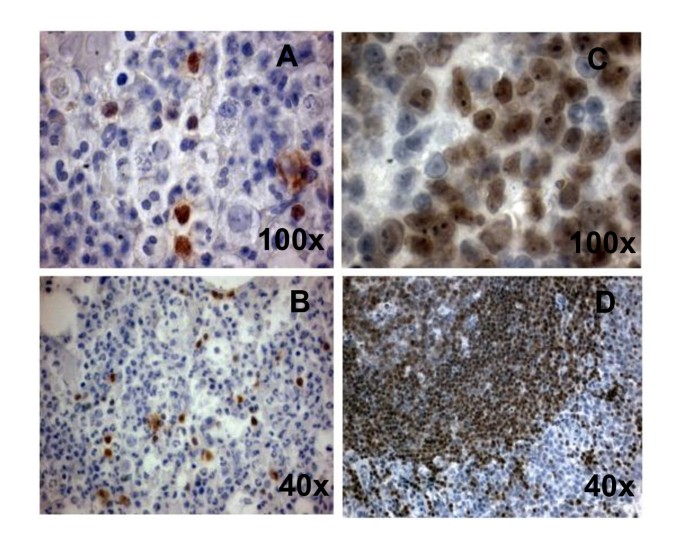
PAX5 expression in bone marrow from pro-B ALL at AML remission. Immunohistochemistry for PAX5 was performed on a marrow biopsy. The frequency of PAX5 cells was 15% (A, B). PAX5 expression by lymph node germinal center cells is shown for control (C, D).
Full size image
Briefly, one marrow biopsies fixed in Bouin’s solution were decalcified, and paraffin-embedded tissue sections were stained with an antibody against the PAX5. The immunocytochemistry revealed scanty PAX5 expression. (Only 15% of cells in the specimen were positive). The same test was not conducted at relapse because no autopsy was performed.
Discussion
Lineage commitment and differentiation to a mature cell type are considered to be unidirectional and irreversible processes under physiological conditions. However, a number of studies with murine models suggest that plasticity is a significant attribute of hematopoietic progenitors, and the process of cell formation is much more dynamic than previously thought [11]. Lineage switching is an example of the lineage heterogeneity that exists in some acute leukemias [12]. The frequency of lineage switching among patients with acute leukemia that relapse is estimated to be between 6% and 9% [6].
Some conditions could explain the lineage change at relapse, being one of them the possibility of a second neoplasm, following two or three years of high dose etoposide treatment[13]. Our patient received 1,700 mg/m2 of etoposide in the first chemotherapy cycle, and the relapse occurred only 2 months after treatment. It is less probable to develop a secondary leukemia before a latency period of two years. Another hypothesis to explain immunophenotype changes at relapse is clonal selection. This mechanism has been proposed in patients with a persistent TEL-AML1+ preleukemic/leukemic clone at relapse [14].
We performed karyotype analysis for our patient and did not find any cytogenetic alteration. In some instances, an acute leukemia lineage switch may represent the emergence of a new leukemic clone characterized by different morphology than the observed upon initial diagnosis, without gene rearrangements upon conversion to another lineage. Zardo et al. propose that the plasticity and reversibility of modifications affecting chromatin structure are important in the expression of genes involved in cell fate decisions and in maintenance of cell-differentiated states. Epigenetic changes in DNA and chromatin, which must occur to allow accessibility to transcription factors at specific DNA-binding sites, are regarded as emerging major players for hematopoietic stem cell lineage differentiation [15].
Mantadakis et al. [16] described the in vivo conversion of T-cell acute lymphoblastic leukemia (ALL), with an early thymocyte immunophenotype and no myeloid markers, to acute myeloid leukemia (AML). This evidence suggests that a subset of T-cell leukemias with minimal differentiation can display a relapse as another lineage, AML.
Palomero et al. suggest that the leukemic transformation may occur in early progenitors and might be influenced by external and internal clues; they propose mutations in the NOTCH1 transcription factor as responsible in lineage switch leukemias [17].
In our patient, lineage markers revealed the emergence of apparently new lymphoid populations. In this case the new ‘relapse lymphoid clone’ was not detected at the moment of surveillance in between the first and secondary diagnosis by investigation of the B related transcription factor PAX5. The selection and emergence of chemo-resistant sub-clones that are undetectable by routine methods, or selection of a new leukemic clone have been previously reported by Henderson et al [18], and we can not ruled out either of this possibilities for our patient.
Studies of normal blood cell development and malignant transformation of hematopoietic cells have shown that the correctly regulated expression of stage- and lineage-specific genes is a key issue in hematopoiesis. The transcription factor PAX5 has been shown to be a B-cell-specific activator protein and seems to be crucial for B lymphopoiesis, including B-lymphoblastic neoplasms [19, 20].
The immunocytochemistry for PAX5 suggests that at least at the moment of the clinical remission, there was no expression of a transcription factor of lymphoid origin, and between the first and second leukemias, there was a period of time where there were no data supporting lymphoid malignancy until the patient relapsed.
The absence of a lymphoid transcription factor at the beginning of surveillance suggests that the lineage switch occurred upon relapse. These data put forward the possibility of de novo lymphoid leukemia after myeloid leukemia. Further studies on the mechanisms of lineage commitment and differentiation in acute leukemia cases will help us to understand the cell and molecular biology of this phenomenon.
Conclusion
Here, we present one case of lineage switch, from AML to ALL which we consider of great interest due to the importance of early recognition of the specific leukemic lineage, and the medical aid necessary for appropriate treatment. Lineage switching is a rare but well-documented phenomenon. The change in morphology, the cell phenotype at relapse, and the absence of a crucial lymphoid transcription factor when the patient was under clinical surveillance all suggest that a lineage switch from AML to ALL might represent a heterogeneous original clone at the molecular level or the emergence of a second new leukemic clone that lacks molecular heterogeneity.
Consent
Written informed consent was obtained from the patient’s father for publication of this case report and accompanying images. A copy of the written consent is available for review by the Editor-in-Chief of this journal.
References
-
Pui CH, Evans WE: Treatment of acute lymphoblastic leukemia. N Engl J Med. 2006, 354: 166-178. 10.1056/NEJMra052603.
Article
CAS
PubMedGoogle Scholar
-
Smith OP, Hann IM: Clinical features and therapy of lymphoblastic leukemia. Pediatric Hematology. Edited by: Arceci RJ, Hann IM, Smith OP. 2006, Oxford: Blackwell Publishing, 450-481. full_text. 3
Chapter
Google Scholar
-
Krause DS, Van Etten RA: Right on target: eradicating leukemic stem cells. Trends Mol Med. 2007, 13: 470-481.
Article
CAS
PubMed
PubMed CentralGoogle Scholar
-
Harned TM, Gaynon PS: Treating refractory leukemias in childhood, role of clofarabine. Ther Clin Risk Manag. 2008, 4: 327-336.
CAS
PubMed
PubMed CentralGoogle Scholar
-
Golub TR, Arceci RJ: Acute Myelogenous Leukemia. Principles and practice of pediatric oncology. 2006, Lippincott Williams & Wilkins; Philadelphia, 591-644. 5
Google Scholar
-
Gagnon GA, Childs CC, LeMaistre A, Keating M, Cork A, Trujillo JM, Nellis K, Freireich E, Stass SA: Molecular heterogeneity in acute leukemia lineage switch. Blood. 1989, 74: 2088-2095.
CAS
PubMedGoogle Scholar
-
Stass S, Mirro J, Melvin S, Pui CH, Murphy SB, Williams D: Lineage switch in acute leukemia. Blood. 1984, 64: 701-706.
CAS
PubMedGoogle Scholar
-
Stass SA, Mirro J: Lineage heterogeneity in acute leukemia: Acute mixed-lineage leukemia and lineage switch. Clin Haematol. 1986, 15: 811-827.
CAS
PubMedGoogle Scholar
-
Zarrabi MH, Rosner F, Grunwald HW: Second neoplasms in acute lymphoblastic leukemia. Cancer. 1983, 52: 1712-1719. 10.1002/1097-0142(19831101)52:9<1712::AID-CNCR2820520927>3.0.CO;2-I.
Article
CAS
PubMedGoogle Scholar
-
Cobaleda C, Schebasteda A, Delogu A, Busslinges M: PAX5: the guardian of B cell identity and function. Nat Immunol. 2007, 8: 463-470. 10.1038/ni1454.
Article
CAS
PubMedGoogle Scholar
-
Welner RS, Pelayo R, Kincade PW: Envolving views on the genealogy of B cells. Evolving views on the genealogy of B cells. Nat Rev Immunol. 2008, 8: 95-10.1038/nri2234.
Article
CAS
PubMedGoogle Scholar
-
Hurwitz CA, Mirro J: Mixed-lineage leukemia and asynchronous antigen expression. Hematol Oncol Clin North Am. 1990, 4: 767-794.
CAS
PubMedGoogle Scholar
-
Tolcher AW, Rowinsky EK: DNA Topoisomerase II inhibitors. The Chemotherapy Source Book. Edited by: Perry MC. 2001, Philadelphia: Lippicott Williams and Wilkins, 278-289. 3
Google Scholar
-
Panzer GE, Cazzaniga G, Velden van der V, Giudice L, Peham M, GeorgMann , Eckert C, Schrauder A, Germano G, Harbott J, Basso G, Biondi A, van Dongen J, Gadner H, Haas O: Immunogenotype Changes Prevail in Relapses of Young Children with TEL-AML1-Positive Acute Lymphoblastic Leukemia and Derive Mainly from Clonal Selection. Clin Cancer Res. 2005, 11: 7719-7727.
Google Scholar
-
Zardo G, Cimino G, Nervi C: Epigenetic plasticity of chromatin in embryonic and hematopoietic stem/progenitor cells: therapeutic potential of cell reprogramming. Leukemia. 2008, 22: 1503-1518. 10.1038/leu.2008.141.
Article
CAS
PubMedGoogle Scholar
-
Mantadakis E, Danilatou V, Stiakaki E, Paterakis G, Papadhimitriou S, Kalmanti M: T-Cell Acute Lymphoblastic Leukemia Relapsing as Acute Myelogenous Leukemia. Ped Blood Cancer. 2007, 48: 354-357. 10.1002/pbc.20543.
Article
Google Scholar
-
Palomero T, McKenna K, O-Neil J, Galinsky I, Stone R, Suzukawa K, Stiakaki E, Kalmanti M, Fox EA, Caligiuri MA, Aster JC, Look AT, Ferrando AA: Activating mutations in NOTCH1 in acute myeloid leukemia and lineage switch leukemias. Leukemia. 2006, 20: 1963-1966. 10.1038/sj.leu.2404409.
Article
CAS
PubMedGoogle Scholar
-
Henderson M, Choi S, Beesley AH, Sutton R, Venn NC, Marshall GM, Kees UR, Haber M, Norris MD: Mechanism of relapse in pediatric acute lymphoblastic leukemia. Cell Cycle. 2008, 7 (10): 1315-1320.
Article
CAS
PubMedGoogle Scholar
-
Falini B, Mason DY: Proteins encoded by genes involved in chromosomal alterations in lymphoma and leukemia: clinical value of their detection by immunocytochemistry. Blood. 2002, 99: 409-426. 10.1182/blood.V99.2.409.
Article
CAS
PubMedGoogle Scholar
-
Smith E, Sigvardsson M: The roles of transcription factors in B lymphocyte commitment, development, and transformation. J Leukoc Biol. 2004, 75: 973-981. 10.1189/jlb.1103554.
Article
CAS
PubMedGoogle Scholar
Download references
Author information
Authors and Affiliations
-
Department of Pediatric Hematology and Oncology, Hospital Infantil de Mexico Federico Gomez, Mexico City, Mexico
Elisa Dorantes-Acosta, Farina Arreguin-Gonzalez & Aurora Medina-Sanson
-
Department of Critical Care Medicine, Instituto Nacional de Ciencias Medicas y Nutricion Salvador Zubiran, Mexico City, Mexico
Carlos A Rodriguez-Osorio
-
Department of Pathology, Hospital Infantil de Mexico Federico Gomez, Mexico City, Mexico
Stanislaw Sadowinski
-
Oncology Research Unit, Oncology Hospital, National Medical Center, IMSS, Mexico City, Mexico
Rosana Pelayo
Authors
- Elisa Dorantes-Acosta
You can also search for this author in
PubMed Google Scholar - Farina Arreguin-Gonzalez
You can also search for this author in
PubMed Google Scholar - Carlos A Rodriguez-Osorio
You can also search for this author in
PubMed Google Scholar - Stanislaw Sadowinski
You can also search for this author in
PubMed Google Scholar - Rosana Pelayo
You can also search for this author in
PubMed Google Scholar - Aurora Medina-Sanson
You can also search for this author in
PubMed Google Scholar
Corresponding author
Correspondence to
Elisa Dorantes-Acosta.
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
EDA analyzed and interpreted the patient data regarding the hematological disease and the relapse. SS performed the histological examination of the bone marrow. EDA, FAG, CARO, RP, and AMS were major contributors in writing the manuscript and analyzing the results. All authors read and approved the final manuscript.
Authors’ original submitted files for images
Rights and permissions
This article is published under license to BioMed Central Ltd. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Reprints and Permissions
About this article
Cite this article
Dorantes-Acosta, E., Arreguin-Gonzalez, F., Rodriguez-Osorio, C.A. et al. Acute myelogenous leukemia switch lineage upon relapse to acute lymphoblastic leukemia: a case report.
Cases Journal 2, 154 (2009). https://doi.org/10.1186/1757-1626-2-154
Download citation
-
Received: 06 January 2009
-
Accepted: 15 October 2009
-
Published: 15 October 2009
-
DOI: https://doi.org/10.1186/1757-1626-2-154
Keywords
- Acute Myeloid Leukemia
- Acute Lymphoblastic Leukemia
- Cytarabine
- Acute Leukemia
- Blast Cell
- Case Report
- Open Access
- Published: 15 October 2009
- Farina Arreguin-Gonzalez1,
- Carlos A Rodriguez-Osorio2,
- Stanislaw Sadowinski3,
- Rosana Pelayo4 &
- …
- Aurora Medina-Sanson1
Cases Journal
volume 2, Article number: 154 (2009)
Cite this article
-
7171 Accesses
-
9 Citations
-
Metrics details
Abstract
Acute leukemia, the most common form of cancer in children, accounts for approximately 30% of all childhood malignancies, with acute lymphoblastic leukemia being five times more frequent than acute myeloid leukemia. Lineage switch is the term that has been used to describe the phenomenon of acute leukemias that meet the standard French-American-British system criteria for a particular lineage (either lymphoid or myeloid) upon initial diagnosis, but meet the criteria for the opposite lineage at relapse. Many reports have documented conversions of acute lymphoblastic leukemia to acute myeloid leukemia.
Here, we report the case of a 4-year-old child with acute myeloid leukemia, which upon relapse switched to acute lymphoblastic leukemia. The morphologic, phenotypic, and molecular features suggest the origin of a new leukemic clone.
Introduction
Leukemia is a group of malignant diseases of the hematopoietic system characterized by the uncontrolled overproduction of either immature (acute leukemia) or terminally differentiated (chronic leukemia) leukocytes.
Acute leukemia, the most common form of cancer in children, accounts for approximately 30% of all childhood malignancies [1]. A frequency of ≥ 20% blasts is required to confirm a diagnosis of acute leukemia. The first comprehensive morphologic classification system for acute leukemias was the French-American-British (FAB) classification, which was established in 1976 and revised in 1985. Other recommended tests are immunophenotyping, cytogenetics, and molecular genetics tests [2].
Acute leukemias can be further sub classified by determining whether the malignant leukocytes are of myeloid origin (cells of granulocyte, monocyte, erythroid, or megakaryocyte lineage) or lymphoid origin (B-cells, T-cells) [3].
Overall survival for patients with acute lymphoblastic leukemia is approximately 80%; currently for acute myeloid leukemia (AML), most large studies show a five-year event-free-survival rate of almost 50%, despite the fact that between one third and one half of patients with AML experience relapse, and no standard therapy is recognized for patients with relapsed and/or refractory AML. Relapses mostly exhibit the same FAB subtype. Patients with relapsed/refractory AML usually respond less well and for a shorter duration to reinduction therapies. This clearly points to the induction of drug-resistant mechanisms and is, in part, related to specific chromosomal abnormalities and the duration of the first remission [4, 5].
However, unexplained variability in clinical course still exists among some individuals within defined risk-group strata.
«Lineage switch» is the term that has been used to describe the phenomenon of acute leukemias that meet standard FAB criteria for a lineage (lymphoid or myeloid) at initial diagnosis but upon relapse meet the criteria for the opposite lineage [6].
Most reports of lineage switching have demonstrated acute lymphoblastic leukemia (ALL) to acute myeloid leukemia (AML) conversions [7–9]. The prognosis for these patients is variable, and there is no standard treatment for them.
Case presentation
A 4-year-old Mexican Mestizo (Hispanic) male was admitted to the Hospital Infantil de México Federico Gómez presenting a 30-day history of fever, pallor, and enlarged cervical lymph nodes.
The initial laboratory findings revealed a white blood cell count of 4,300/mm3, with 16% blasts in the peripheral blood, 8.8 g/dL hemoglobin, and a platelet count of 49,000/mm3.
Cerebrospinal fluid was negative for blast cells. The bone marrow was hypercellular, with 64% blasts, which were large in size and showed low nucleus: cytoplasm ratio, monocytoid features, and blue-gray cytoplasm; > 80% of the blast cells were monoblasts, and the rest were promonocytes or monocytes (Figure 1). The surface immunophenotype was CD45+ (91.1%), CD15+ (95.4%), CD14-/+ (37.7%), CD13+/- (78.5%), HLA-DR+/- (78.5%), CD22-/+ (42.8%), CD10— (2.1%) CD19— (2.5%) CD20— (0.7%) CD7— (0.07%), CD3-/+ (19.4%), and CD33— (7.9%) (Figure 2).
Bone marrow aspiration at AML diagnosis. Blast cells were large in size and had a low nucleus:cytoplasm ratio, monocytoid aspect, and blue-gray cytoplasm. Wright stain, 1000×.
Full size image
Flow cytometry AML at diagnosis. Immunophenotyping of leukemic cells by flow cytometric analyses revealed myeloid blasts negative for CD10 and CD19 (A) but positive for CD13 (B) and CD14 (C).
Full size image
Cytochemistry was positive for myeloperoxidase (MPO). Conventional cytogenetic analysis using direct and 24-hour unstimulated and unsynchronized cultures revealed a 46 XY karyotype. A diagnosis of M5 AML was made. The patient started chemotherapy with a modified MRC-10 protocol: daunorubicin 50 mg/m2 (days 1, 3 & 5); cytarabine 100 mg/m2 in a 1-hour infusion every 12 hours through days 1 to 10; and etoposide 100 mg/m2 in a 4-hour infusion every 24 hours through days 1 to 5.
Due to oral candidiasis and an episode of sepsis secondary to Acinetobacter iwofii, the patient was treated with meropenem, amphotericin, and fluconazole.
Remission was determined by marrow aspiration with no blast cells at day 27th after first chemotherapy cycle.
Thirty days after the first course of chemotherapy, a second chemotherapy cycle was administered without complications daunorubicin 50 mg/m2 days 1, 3 & 5; cytarabine 100 mg/m2 in a 1-hour infusion every 12 hours through days 1 to 4; and etoposide 100 mg/m2 in a 4-hour infusion every 24 hours through days 1 to 5.
A third cycle: etoposide 100 mg/m2 in a 4-hour infusion every 24 hours on days 1-5 and cytarabine 1000 mg/m2 in a 1-hour infusion every 12 hours (days 1-3) was stopped on day 3 because of a life-threatening episode of pneumonia; the patient was transferred to the ICU and supported by mechanical ventilation. Because a lung CT scan showed features of a right lung abscess, a medium and right lobectomy was performed.
Upon clinical and hematologic recovery, the patient received a fourth cycle of chemotherapy with mitoxantrone 10 mg/m2 in a 1-hour infusion every 24 hours (days 1-5) and cytarabine 1000 mg/m2 in a 1-hour infusion every 12 hours (days 1-3). Two weeks later, an episode of fever, along with neutropenia and mucositis, was recorded, and the patient was treated with cefepime and amikacin.
The patient received a fifth cycle or chemotherapy: etoposide 100 mg/m2 in a 4-hour infusion every 24 hours for 5 days and cytarabine 1000 mg/m2 in a 1-hour infusion every 12 hours for 3 days.
Seven months later, after the diagnosis, the chemotherapy protocol was completed with a clearance of blast cells.
Two months later, the patient presented at the hospital with petechiae. Initial laboratory data revealed a white blood cell count of 7,700/mm3, 13% blast cells, 13.7 g/dL hemoglobin, and a platelet count of 33,000/mm3. The marrow was hypercellular and contained 85% blasts. In contrast to the previous neoplasm, the blasts in the current specimen were smaller, with a predominance of small cells, scanty cytoplasm, moderate cytoplasmic basophilia, and variable cytoplasmic vacuolation (Figure 3), along with the immunophenotype CD45+ (81.6%), CD10+ (77%), CD19+ (89%), CD22— (0%), CD20— (3.7%), CD15— (0%), CD14— (4.5%), CD7— (7.8%), CD3— (13.1%), CD13— (0.27%), CD33— (0.62%), and HLA-DR— (0.2%) (Figure 4). These lymphoblasts were negative for myeloperoxidase (data not shown).
Bone marrow aspiration at relapse. Blasts were smaller, with a predominance of small cells, scanty cytoplasm, moderate cytoplasmic basophilia, and variable cytoplasmic vacuolation. Wright stain, 1000×.
Full size image
Flow cytometry at relapse. Flow cytometric analyses of leukemic cells at relapse. Blast cells were identified as lymphoblasts because the immunophenotype was positive for CD10 and CD19 (A) but negative for CD13 and CD14 (B, C).
Full size image
The patient was diagnosed with pro-B lymphoblastic leukemia. Cytogenetic studies performed on the marrow aspirate revealed a karyotype of 46 XY.
We tried to induce remission with the national protocol for acute lymphoblastic leukemias, adapted from TOTXV protocol from St Jude Children’s Research Hospital with vincristine 2 mg/m2 once a week for four weeks, daunorubicin 30 mg/m2 once a week for two weeks, L-asparaginase 10,000 IU/m2 three times a week for a total of nine doses, and dexamethasone 6 mg/m2 for four weeks. However, the bone marrow examination on the 14th and 21th days of induction showed persisting infiltration with 50% blasts, and the patient remained pancytopenic. After an episode of fever, abdominal pain, and diarrhea, antibiotic therapy was conducted.
As the bone marrow examination showed persisting infiltration, we indicated cycle 1 and 2 to myeloid leukemia (described above). This scheme of chemotherapy is used in our hospital for patients with early relapse or refractory acute leukemia. After that, the patient had infectious complications, followed by development of tumor lysis syndrome and septic shock. Chemotherapy was stopped and the patient was transferred to the ICU and supported by mechanical ventilation. After clinical recovery, the bone marrow showed persisting infiltration with 85% blasts, and the patient’s parents asked for palliative care; the child was cared for at home for four weeks under the surveillance of a palliative care clinic. Despite supportive measures, he died six months after the diagnosis of relapse. No autopsy was authorized by the family.
In order to investigate if a B lymphoid transcription factor was already expressed at the time of remission of the myeloid leukemia, we performed detection of PAX5 (B-cell-specific activator protein for B cells, including B-lymphoblastic neoplasms) [10], in bone marrow biopsy taken at the end of the first scheme of chemotherapy, when the patient started surveillance (Figure 5).
PAX5 expression in bone marrow from pro-B ALL at AML remission. Immunohistochemistry for PAX5 was performed on a marrow biopsy. The frequency of PAX5 cells was 15% (A, B). PAX5 expression by lymph node germinal center cells is shown for control (C, D).
Full size image
Briefly, one marrow biopsies fixed in Bouin’s solution were decalcified, and paraffin-embedded tissue sections were stained with an antibody against the PAX5. The immunocytochemistry revealed scanty PAX5 expression. (Only 15% of cells in the specimen were positive). The same test was not conducted at relapse because no autopsy was performed.
Discussion
Lineage commitment and differentiation to a mature cell type are considered to be unidirectional and irreversible processes under physiological conditions. However, a number of studies with murine models suggest that plasticity is a significant attribute of hematopoietic progenitors, and the process of cell formation is much more dynamic than previously thought [11]. Lineage switching is an example of the lineage heterogeneity that exists in some acute leukemias [12]. The frequency of lineage switching among patients with acute leukemia that relapse is estimated to be between 6% and 9% [6].
Some conditions could explain the lineage change at relapse, being one of them the possibility of a second neoplasm, following two or three years of high dose etoposide treatment[13]. Our patient received 1,700 mg/m2 of etoposide in the first chemotherapy cycle, and the relapse occurred only 2 months after treatment. It is less probable to develop a secondary leukemia before a latency period of two years. Another hypothesis to explain immunophenotype changes at relapse is clonal selection. This mechanism has been proposed in patients with a persistent TEL-AML1+ preleukemic/leukemic clone at relapse [14].
We performed karyotype analysis for our patient and did not find any cytogenetic alteration. In some instances, an acute leukemia lineage switch may represent the emergence of a new leukemic clone characterized by different morphology than the observed upon initial diagnosis, without gene rearrangements upon conversion to another lineage. Zardo et al. propose that the plasticity and reversibility of modifications affecting chromatin structure are important in the expression of genes involved in cell fate decisions and in maintenance of cell-differentiated states. Epigenetic changes in DNA and chromatin, which must occur to allow accessibility to transcription factors at specific DNA-binding sites, are regarded as emerging major players for hematopoietic stem cell lineage differentiation [15].
Mantadakis et al. [16] described the in vivo conversion of T-cell acute lymphoblastic leukemia (ALL), with an early thymocyte immunophenotype and no myeloid markers, to acute myeloid leukemia (AML). This evidence suggests that a subset of T-cell leukemias with minimal differentiation can display a relapse as another lineage, AML.
Palomero et al. suggest that the leukemic transformation may occur in early progenitors and might be influenced by external and internal clues; they propose mutations in the NOTCH1 transcription factor as responsible in lineage switch leukemias [17].
In our patient, lineage markers revealed the emergence of apparently new lymphoid populations. In this case the new ‘relapse lymphoid clone’ was not detected at the moment of surveillance in between the first and secondary diagnosis by investigation of the B related transcription factor PAX5. The selection and emergence of chemo-resistant sub-clones that are undetectable by routine methods, or selection of a new leukemic clone have been previously reported by Henderson et al [18], and we can not ruled out either of this possibilities for our patient.
Studies of normal blood cell development and malignant transformation of hematopoietic cells have shown that the correctly regulated expression of stage- and lineage-specific genes is a key issue in hematopoiesis. The transcription factor PAX5 has been shown to be a B-cell-specific activator protein and seems to be crucial for B lymphopoiesis, including B-lymphoblastic neoplasms [19, 20].
The immunocytochemistry for PAX5 suggests that at least at the moment of the clinical remission, there was no expression of a transcription factor of lymphoid origin, and between the first and second leukemias, there was a period of time where there were no data supporting lymphoid malignancy until the patient relapsed.
The absence of a lymphoid transcription factor at the beginning of surveillance suggests that the lineage switch occurred upon relapse. These data put forward the possibility of de novo lymphoid leukemia after myeloid leukemia. Further studies on the mechanisms of lineage commitment and differentiation in acute leukemia cases will help us to understand the cell and molecular biology of this phenomenon.
Conclusion
Here, we present one case of lineage switch, from AML to ALL which we consider of great interest due to the importance of early recognition of the specific leukemic lineage, and the medical aid necessary for appropriate treatment. Lineage switching is a rare but well-documented phenomenon. The change in morphology, the cell phenotype at relapse, and the absence of a crucial lymphoid transcription factor when the patient was under clinical surveillance all suggest that a lineage switch from AML to ALL might represent a heterogeneous original clone at the molecular level or the emergence of a second new leukemic clone that lacks molecular heterogeneity.
Consent
Written informed consent was obtained from the patient’s father for publication of this case report and accompanying images. A copy of the written consent is available for review by the Editor-in-Chief of this journal.
References
-
Pui CH, Evans WE: Treatment of acute lymphoblastic leukemia. N Engl J Med. 2006, 354: 166-178. 10.1056/NEJMra052603.
Article
CAS
PubMedGoogle Scholar
-
Smith OP, Hann IM: Clinical features and therapy of lymphoblastic leukemia. Pediatric Hematology. Edited by: Arceci RJ, Hann IM, Smith OP. 2006, Oxford: Blackwell Publishing, 450-481. full_text. 3
Chapter
Google Scholar
-
Krause DS, Van Etten RA: Right on target: eradicating leukemic stem cells. Trends Mol Med. 2007, 13: 470-481.
Article
CAS
PubMed
PubMed CentralGoogle Scholar
-
Harned TM, Gaynon PS: Treating refractory leukemias in childhood, role of clofarabine. Ther Clin Risk Manag. 2008, 4: 327-336.
CAS
PubMed
PubMed CentralGoogle Scholar
-
Golub TR, Arceci RJ: Acute Myelogenous Leukemia. Principles and practice of pediatric oncology. 2006, Lippincott Williams & Wilkins; Philadelphia, 591-644. 5
Google Scholar
-
Gagnon GA, Childs CC, LeMaistre A, Keating M, Cork A, Trujillo JM, Nellis K, Freireich E, Stass SA: Molecular heterogeneity in acute leukemia lineage switch. Blood. 1989, 74: 2088-2095.
CAS
PubMedGoogle Scholar
-
Stass S, Mirro J, Melvin S, Pui CH, Murphy SB, Williams D: Lineage switch in acute leukemia. Blood. 1984, 64: 701-706.
CAS
PubMedGoogle Scholar
-
Stass SA, Mirro J: Lineage heterogeneity in acute leukemia: Acute mixed-lineage leukemia and lineage switch. Clin Haematol. 1986, 15: 811-827.
CAS
PubMedGoogle Scholar
-
Zarrabi MH, Rosner F, Grunwald HW: Second neoplasms in acute lymphoblastic leukemia. Cancer. 1983, 52: 1712-1719. 10.1002/1097-0142(19831101)52:9<1712::AID-CNCR2820520927>3.0.CO;2-I.
Article
CAS
PubMedGoogle Scholar
-
Cobaleda C, Schebasteda A, Delogu A, Busslinges M: PAX5: the guardian of B cell identity and function. Nat Immunol. 2007, 8: 463-470. 10.1038/ni1454.
Article
CAS
PubMedGoogle Scholar
-
Welner RS, Pelayo R, Kincade PW: Envolving views on the genealogy of B cells. Evolving views on the genealogy of B cells. Nat Rev Immunol. 2008, 8: 95-10.1038/nri2234.
Article
CAS
PubMedGoogle Scholar
-
Hurwitz CA, Mirro J: Mixed-lineage leukemia and asynchronous antigen expression. Hematol Oncol Clin North Am. 1990, 4: 767-794.
CAS
PubMedGoogle Scholar
-
Tolcher AW, Rowinsky EK: DNA Topoisomerase II inhibitors. The Chemotherapy Source Book. Edited by: Perry MC. 2001, Philadelphia: Lippicott Williams and Wilkins, 278-289. 3
Google Scholar
-
Panzer GE, Cazzaniga G, Velden van der V, Giudice L, Peham M, GeorgMann , Eckert C, Schrauder A, Germano G, Harbott J, Basso G, Biondi A, van Dongen J, Gadner H, Haas O: Immunogenotype Changes Prevail in Relapses of Young Children with TEL-AML1-Positive Acute Lymphoblastic Leukemia and Derive Mainly from Clonal Selection. Clin Cancer Res. 2005, 11: 7719-7727.
Google Scholar
-
Zardo G, Cimino G, Nervi C: Epigenetic plasticity of chromatin in embryonic and hematopoietic stem/progenitor cells: therapeutic potential of cell reprogramming. Leukemia. 2008, 22: 1503-1518. 10.1038/leu.2008.141.
Article
CAS
PubMedGoogle Scholar
-
Mantadakis E, Danilatou V, Stiakaki E, Paterakis G, Papadhimitriou S, Kalmanti M: T-Cell Acute Lymphoblastic Leukemia Relapsing as Acute Myelogenous Leukemia. Ped Blood Cancer. 2007, 48: 354-357. 10.1002/pbc.20543.
Article
Google Scholar
-
Palomero T, McKenna K, O-Neil J, Galinsky I, Stone R, Suzukawa K, Stiakaki E, Kalmanti M, Fox EA, Caligiuri MA, Aster JC, Look AT, Ferrando AA: Activating mutations in NOTCH1 in acute myeloid leukemia and lineage switch leukemias. Leukemia. 2006, 20: 1963-1966. 10.1038/sj.leu.2404409.
Article
CAS
PubMedGoogle Scholar
-
Henderson M, Choi S, Beesley AH, Sutton R, Venn NC, Marshall GM, Kees UR, Haber M, Norris MD: Mechanism of relapse in pediatric acute lymphoblastic leukemia. Cell Cycle. 2008, 7 (10): 1315-1320.
Article
CAS
PubMedGoogle Scholar
-
Falini B, Mason DY: Proteins encoded by genes involved in chromosomal alterations in lymphoma and leukemia: clinical value of their detection by immunocytochemistry. Blood. 2002, 99: 409-426. 10.1182/blood.V99.2.409.
Article
CAS
PubMedGoogle Scholar
-
Smith E, Sigvardsson M: The roles of transcription factors in B lymphocyte commitment, development, and transformation. J Leukoc Biol. 2004, 75: 973-981. 10.1189/jlb.1103554.
Article
CAS
PubMedGoogle Scholar
Download references
Author information
Authors and Affiliations
-
Department of Pediatric Hematology and Oncology, Hospital Infantil de Mexico Federico Gomez, Mexico City, Mexico
Elisa Dorantes-Acosta, Farina Arreguin-Gonzalez & Aurora Medina-Sanson
-
Department of Critical Care Medicine, Instituto Nacional de Ciencias Medicas y Nutricion Salvador Zubiran, Mexico City, Mexico
Carlos A Rodriguez-Osorio
-
Department of Pathology, Hospital Infantil de Mexico Federico Gomez, Mexico City, Mexico
Stanislaw Sadowinski
-
Oncology Research Unit, Oncology Hospital, National Medical Center, IMSS, Mexico City, Mexico
Rosana Pelayo
Authors
- Elisa Dorantes-Acosta
You can also search for this author in
PubMed Google Scholar - Farina Arreguin-Gonzalez
You can also search for this author in
PubMed Google Scholar - Carlos A Rodriguez-Osorio
You can also search for this author in
PubMed Google Scholar - Stanislaw Sadowinski
You can also search for this author in
PubMed Google Scholar - Rosana Pelayo
You can also search for this author in
PubMed Google Scholar - Aurora Medina-Sanson
You can also search for this author in
PubMed Google Scholar
Corresponding author
Correspondence to
Elisa Dorantes-Acosta.
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
EDA analyzed and interpreted the patient data regarding the hematological disease and the relapse. SS performed the histological examination of the bone marrow. EDA, FAG, CARO, RP, and AMS were major contributors in writing the manuscript and analyzing the results. All authors read and approved the final manuscript.
Authors’ original submitted files for images
Rights and permissions
This article is published under license to BioMed Central Ltd. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Reprints and Permissions
About this article
Cite this article
Dorantes-Acosta, E., Arreguin-Gonzalez, F., Rodriguez-Osorio, C.A. et al. Acute myelogenous leukemia switch lineage upon relapse to acute lymphoblastic leukemia: a case report.
Cases Journal 2, 154 (2009). https://doi.org/10.1186/1757-1626-2-154
Download citation
-
Received: 06 January 2009
-
Accepted: 15 October 2009
-
Published: 15 October 2009
-
DOI: https://doi.org/10.1186/1757-1626-2-154
Keywords
- Acute Myeloid Leukemia
- Acute Lymphoblastic Leukemia
- Cytarabine
- Acute Leukemia
- Blast Cell
Хронический миелоидный лейкоз – это онкологическое заболевание, приводящее к образованию избыточного количества измененных клеток крови и накоплению их в различных тканях и органах.

Как возникает миелолейкоз?
Все здоровые клетки в теле человека действуют по «программе», определяемой генами – рождаются, работают и умирают. Периодически во время их деления возникают сбои и вместо одной нормальной клетки в организме появляется две измененных. Это происходит довольно часто, но большинство из них выявляет и уничтожает иммунная система.
Иногда они образуются в костном мозге – мягкой пористой сердцевине некоторых крупных костей, в которой содержатся миелоидные клетки, производящие различные клетки крови:
- эритроциты, переносящие кислород из легких к тканям тела и возвращающие в дыхательные пути углекислый газ;
- большую часть видов лейкоцитов, противостоящих инфекциям;
- и тромбоциты, необходимые для прекращения кровотечений.
Если естественной защите организма не удается справиться со своей задачей, измененные миелоидные клетки превращаются в лейкемические и приобретают крайне опасные свойства:
- они не созревают и не производят обычные эритроциты, лейкоциты и тромбоциты;
- быстро развиваются и размножаются;
- игнорируют апоптоз – механизм «запрограммированной» смерти, и не погибают по окончанию нормального жизненного цикла;
- накапливаются в костном мозге и вытесняют из него нормальные клетки;
- со временем покидают его и попадают в кровоток;
- проникают в разные части тела;
- скапливаются в некоторых органах, таких как селезенка;
- и нарушают работу других клеток и тканей.
Хронический миелоидный лейкоз развивается довольно медленно, а многие его обладатели на протяжении длительного времени не подозревают о наличии у себя заболевания и не ощущают его симптомов.
Лейкоз крови: лечение и прогнозы.
Костный мозг и кровь
Лейкоз – это сложное для понимания заболевание. Для того, чтобы разобраться в его сути, необходимо изучить некоторую информацию о кровеносной и кроветворной системах.
Костный мозг – это мягкая, пористая внутренняя часть некоторых костей, таких как ребра, таз, позвоночник, череп и лопатки. Он состоит из:
- небольшого количества стволовых клеток, из которых образуются различные клетки крови;
- жировых клеток;
- соединительных – поддерживающих тканей, которые помогают клеткам расти.
Типы клеток крови:
-
Красные кровяные тельца, или эритроциты
– переносят кислород к тканям и забирают из них углекислый газ. - Тромбоциты – это оторвавшиеся фрагменты содержащихся в костном мозге мегакариоцитов. Закупоривают стенки сосудов и останавливают кровотечения.
- Белые кровяные тельца, или лейкоциты – помогают организму противостоять инфекциям.
Типы лейкоцитов:
Лимфоциты
– составляют основную ткань иммунной системы, защищают тело от микробов и регулируют работу иммунитета.
Гранулоциты
– содержат вещества, которые могут уничтожать вредные микроорганизмы, такие как бактерии.
Моноциты
– распознают, окружают и уничтожают микробов.

Симптомы и признаки хронического миелоидного лейкоза
Проявления лейкоза могут быть похожи на множество различных нарушений и заболеваний. Они могут выглядеть как:
- ощущение постоянной усталости;
- боль или чувство переполнения в животе;
- ночная потливость;
- повышение температуры;
- снижение веса;
- боль в костях, вызванную распространением лейкозных клеток из костного мозга на поверхность частей скелета или в суставы;
- увеличение селезенки, расположенной слева, в верхней части брюшной полости, позади желудка;
- ощущение сытости после приема небольшого количества пищи.
Многие симптомы возникают из-за замещения нормальных кроветворных клеток костного мозга лейкозными, что вызывает:
- Анемию – дефицит переносящих кислород эритроцитов, а следовательно – слабость, усталость и одышку, или ощущение нехватки воздуха.
- Лейкопению – нехватку нормальных лейкоцитов и частое развитие инфекций.
- Нейтропению – отсутствие достаточного количества нейтрофилов, вызывающее тяжелые бактериальные заболевания.
- Тромбоцитопению – низкий уровень тромбоцитов, образование синяков и частые кровотечения, включая носовые.
Один из самых распространенных признаков хронического миелоидного лейкоза – это крайне высокий уровень лейкоцитов в крови.
Почему развивается хронический миелоидный лейкоз?
Нормальные клетки образуются и работают согласно «программе», содержащейся в их хромосомах – наборе переданных родителями генов. Некоторые из них помогают им расти и размножаться. Они называются онкогенами. Другие – супрессоры опухолей – замедляют этот процесс и в нужный момент вызывают их гибель.
В любой момент при делении одной клетки на 2 новых может произойти сбой, приводящий к «включению» первых из них, и «выключению» вторых.
В результате таких ошибок:
- часть двадцать второй хромосомы меняется с небольшой областью девятой;
- она укорачивается и превращается в так называемую филадельфийскую хромосому, которая обнаруживается в лейкозных клетках почти всех пациентов;
- образуется особый онкоген BCR-ABL;
- и в организме развивается лейкоз.
Причины подобных сбоев на сегодняшний момент не понятны – ученым известно только несколько факторов, повышающих вероятность такого события. К ним относятся:
- Воздействие высокой дозы радиации, полученной в результате взрыва или аварии.
- Возраст: чем старше человек, чем выше его риски.
- Пол: у мужчин заболевание встречается немного чаще, чем у женщин.
По имеющимся на сегодняшний день научным данным, на шансы развития хронического миелоидного лейкоза не влияют курение, воздействие других химических веществ, питание и инфекции. Кроме того, он не передается по наследству – все связанные с ним изменения в генах возникают в течение жизни.

Диагностика хронического миелоидного лейкоза
Заболевание часто обнаруживается случайно, когда человек сдает анализы для выявления причин не связанных с ним проблем со здоровьем или во время плановой диспансеризации. Если у специалистов появляется повод подозревать наличие лейкоза, они назначают дополнительные исследования:
Общий анализ: измерение количества различных клеток крови – переносящих кислород эритроцитов, противостоящих инфекциям лейкоцитов и останавливающих кровотечения тромбоцитов. Кроме того, с его помощью определяют процент измененных клеток, не способных выполнять свои задачи.
Анализ костного мозга – важная составляющая процедуры диагностики, поскольку именно в этом органе начинает развиваться лейкоз. Его образцы получают в ходе 2 процедур, которые обычно назначаются одновременно:
- Для выполнения аспирации человек ложится на бок или на живот, врач очищает и обезболивает кожу в области бедра, после чего вводит тонкую полую иглу в кость и отсасывает ее жидкое содержимое.
- Как правило, затем проводится биопсия – изъятие небольшого кусочка кости и костного мозга с помощью специального инструмента.
Затем специалисты подсчитывают количество кроветворных клеток в материалах. У людей с миелолейкозом их часто оказывается много, поскольку в них содержится большое количество лейкозных клеток.
Кроме того, эти вмешательства необходимы и после постановки диагноза – с помощью их данных оценивается эффективность проводимой терапии.
Биохимический анализ крови: выявляет в ней различные химические вещества и проблемы с печенью или почками, вызванные распространением измененных клеток и побочными эффектами лекарств. Помогает понять, необходим ли человеку прием различных препаратов для корректировки уровней минералов в крови.
Генетические тесты: обнаруживают измененные хромосомы и лейкозный ген BCR-ABL. Используются для подтверждения диагноза и получения дополнительной информации о неправильных клетках.
Компьютерная томография, КТ – создание множества рентгеновских снимков, которые объединяются в одно подробное изображение. Помогает обнаруживать укрупненные лимфатические узлы – «фильтры» организма, задерживающие и обезвреживающие опасные вещества. Назначается при подозрении на поражение селезенки. Кроме того, дает врачам возможность четко направлять иглу при биопсии – взятии образцов тканей.
Магнитно-резонансная томография, МРТ – позволяет изучать состояние мягких внутренних тканей. Особенно эффективна при исследовании головного и спинного мозга.
УЗИ можно использовать для осмотра лимфатических узлов, которые расположены рядом с поверхностью тела, или для выявления увеличенных органов, таких как почки, печень и селезенка.
В онкологическом центре «Лапино-2» выполняется полная диагностика хронического миелоидного лейкоза.
Мы проводим все возможные исследования, берем анализы и быстро получаем их результаты из собственной лаборатории.
Пациентам, которые наблюдаются или проходят лечение в других клиниках, мы предлагаем услугу второго мнения – консультацию опытного врача любой специальности. Наши доктора помогут вам убедиться в правильности имеющихся заключений или выявить в них неточности.
Фазы хронического миелоидного лейкоза
Большинство видов рака классифицируется по стадиям, определяемым согласно размерам опухоли и количеству поврежденных тканей. Такая разбивка позволяет врачам представлять примерные прогнозы развития онкологии и подбирать оптимальное для каждого конкретного случая лечение.
Перспективы пациента с миелолейкозом зависят от других факторов:
- фазы заболевания;
- количества в костном мозге бластов, из которых образуются нормальные клетки крови;
- возраста человека;
- увеличения селезенки – мягкого губчатого органа размером примерно с человеческий кулак, расположенного под грудной клеткой в верхней левой части брюшной полости. Она выполняет множество функций: борется с инфекциями, фильтрует кровь и хранит некоторые ее клетки;
- и данных анализов крови и костного мозга.
Хроническая фаза
Начальный этап, на котором выявляется более 90% всех случаев заболевания.
У таких пациентов обычно есть менее 10% бластов в крови и костном мозге, а симптомы очень легкие или их совсем нет. Они хорошо реагируют на стандартные схемы лечения.
Фаза акселерации
К ней относят все случаи, для которых верно любое из следующих условий:
- в крови более 15%, но менее 30% бластов;
- крайне низкое количество тромбоцитов, уменьшившееся не из-за лечения;
- 20% крови и более составляют базофилы – разновидность лейкоцитов, которые поглощают чужеродные вещества и запускают механизм аллергии – ответа организма на вторжение в него вредных веществ;
- более чем 30% от всего объема крови составляют бласты и промиелоциты, которые превращаются в миелоциты;
- появление новых изменений в хромосомахХромосомы – это часть молекулы ДНК, которая хранит генетический материал человека. лейкозных клеток.
На данном этапе болезни нередко появляются такие симптомы, как повышенная температура, снижение аппетита и потеря веса. Пациенты в фазе акселерации хуже реагируют на лечение.
Бластный криз
Кровь или костный мозг содержат 20% или более бластов. Они скапливаются в этом важном органе, распространяются за его пределы в другие ткани и вызывают проблемы с аппетитом, уменьшение массы тела и лихорадку.

Лечение хронического миелоидного лейкоза
Современная медицина предлагает пациентам несколько вариантов борьбы с заболеванием. Схема лечения подбирается для каждого человека индивидуально. Она может включать в себя:
Таргетную терапию – препараты, воздействующие только на определенные изменения в тканях.
Ген BCR-ABL содержится исключительно в лейкозных клетках и отсутствует в нормальных. Он производит одноименный белок, который помогает им быстро расти и бесконтрольно размножаться. Для противодействия ему созданы специальные препараты, так называемые ингибиторы тирозинкиназы:
- Иматиниб (Гливек);
- Дазатиниб (Спрайсел);
- Нилотиниб (Тасигна);
- Бозутиниб (Бозулиф);
- Понатиниб (Иклюзиг);
- Асциминиб (Скембликс).
Они лучше всего работают, когда хронический миелоидный лейкоз находится в хронической фазе, но могут помочь и на более поздних этапах.
Такие лекарства не излечивают большинство пациентов полностью, из-за чего их приходится принимать постоянно. В части случаев они работают настолько хорошо, что со временем от них удается отказаться или снизить дозу.
Иммунотерапия – усиление работы иммунитета для замедления роста и деления лейкозных клеток. Включает в себя введение интерферонов – искусственно созданных препаратов, действующих как вещество, которое вырабатывается иммунной системой человека. Раньше они применялись как основное лечение, но теперь их назначают редко, так как ингибиторы тирозинкиназы дают больший эффект.
Химиотерапия – препараты, которые доставляются в вену или принимаются внутрь, попадают в кровоток и убивают измененные клетки во всех областях тела. Относительно недавно этот метод был одним из лучших способов борьбы с заболеванием, но в настоящее время он обычно используется в тех случаях, когда Иматиниб и подобные ему средства перестают работать.
Лучевая терапия – уничтожение неправильных клеток радиоактивным облучением. Как правило, назначается для воздействия на увеличенные внутренние органы, такие как селезенка, которые давят на другие ткани, вызывая дискомфорт и другие неприятные симптомы. Кроме того, она применяется для облегчения боли, вызванной повреждением костей при разрастании лейкозных клеток в костном мозге.
Хирургия. Если заболевание поражает селезенку, она укрупняется и воздействует на ближайшие структуры. Когда химиотерапия и облучение не могут ее уменьшить, проводится спленэктомия – удаление органа. Эта процедура не играет никакой роли в собственно лечении хронического миелоидного лейкоза, но снимает тяжелые симптомы и улучшает качество жизни.
Трансплантация стволовых клеток. До появления ингибиторов тирозинкиназы метод применялся часто, теперь же он рекомендуется в ограниченном количестве случаев.
Данная процедура может быть назначена очень молодым пациентам, детям, тем, у кого есть подходящий донор – например, брат или сестра, а также при отсутствии реакции организма на Иматиниб и подобные ему средства. В таких ситуациях сначала вводятся высокие дозы химиопрепаратов, убивающих максимальное количество лейкозных клеток, но повреждающих нормальные ткани костного мозга. Затем выполняется пересадка восстанавливающих их кроветворных клеток.
Трансплантация бывает:
- аутологичной, при проведении которой стволовые клетки забираются у пациента до начала лечения;
- и аллогенной, предполагающей использование материала от подходящего донора.
Лечение хронического миелоидного лейкоза по фазам
Схема терапии подбирается в зависимости от этапа заболевания, возраста человека и других факторов.
Хроническая фаза
Стандартным лечением являются ингибиторы тирозинкиназы – вещества, которые не позволяют особому белку ускорять рост и размножение лейкозных клеток. К ним относятся такие препараты, как Иматиниб, Нилотиниб, Дазатиниб или Бозутиниб. Если первый из них не работает, вызывает тяжелые побочные эффекты или перестает действовать, его дозу увеличивают или назначают следующее средство.
При отсутствии хорошей реакции применяются интерферон или химиотерапия.
Части пациентов может быть назначена трансплантация стволовых клеток.
Фаза акселерации
Если человек ранее не получал никакого лечения, ему предлагается Иматиниб. Большинство людей реагируют на него хорошо, но его эффект длится не так долго, как при хронической фазе. На этом этапе часто используются более новые средства, такие как Дазатиниб и Нилотиниб.
Еще один вариант помощи – это интерферон, но он в этой фазе также работает хуже.
Для большинства молодых пациентов лучшим методом может быть аллогенная трансплантация – пересадка кроветворных клеток от подходящего донора. До ее начала часто проводится химиотерапия.
Бластный криз
Людям, которые не получали ранее никакой терапии, могут быть введены высокие дозы Иматиниба. Дазатиниб, Нилотиниб и Бозутиниб более эффективны на этой стадии, особенно если они еще не применялись. Тем, кто реагирует на данные препараты, возможно назначение трансплантации стволовых клеток.
У части пациентов измененные клетки чувствительны к химиопрепаратам. Примерно в половине таких случаев можно добиться ремиссии, или исчезновения признаков болезни.
Пересадка кроветворных клеток от донора менее эффективна, но она является единственным шансом на полную победу над лейкозом.
На данном этапе важную роль играет паллиативное лечение, направленное не на излечение, а на облегчение симптомов. Например, с помощью лучевой терапии можно уменьшить увеличенную селезенку или снять боль в пораженных костях.
Любое лечение хронического миелоидного лейкоза можно пройти у нас, в онкологическом центре «Лапино-2».
Наши врачи – доктора и кандидаты медицинских наук с большим опытом в области борьбы с онкологическими заболеваниями, выполняют все возможные вмешательства и оказывают любую необходимую помощь.
Мы подбираем терапию индивидуально для каждого человека, помогаем справиться с побочными эффектами, поддерживаем организм и предлагаем максимальный комфорт всем пациентам.
Прогнозы и выживаемость при хроническом миелоидном лейкозе
Перспективы каждого человека индивидуальны и зависят от нескольких факторов – фазы, на которой было обнаружено заболевание, возраста, общего состояния здоровья и реакции на проводимое лечение.
Самым эффективным средством борьбы с хроническим миелоидным лейкозом являются ингибиторы тирозинкиназы – препараты, которые применяются с 2001 года. В связи с этим имеющаяся у медицинского сообщества информация не может считаться полной, но на сегодняшний день уже есть данные о том, что большинство из получавших их людей живы.
Научное исследование о применении Иматиниба показало, что около 90% принимавших его пациентов продолжают наблюдаться у врача через 5 лет с момента начала лечения.
Кроме того, специалистам известны факторы, которые ухудшают прогноз. К ним относятся:
- фаза акселерации или бластного криза;
- увеличение размеров селезенки;
- поражение костей, вызванное активным ростом лейкозных клеток;
- очень высокое или низкое количество тромбоцитов, необходимых для остановки кровотечений;
- возраст от 60 лет;
- избыток в крови базофилов, которые поглощают опасные для организма чужеродные вещества, и эозинофилов, борющихся с вредными микроорганизмами;
- большое количество изменений хромосом лейкозных клеток.
Лейкоз: причины, симптомы, лечение лейкемии
Причины
Симптомы
Классификация
Осложнения
Диагностика
Лечение
")
Лейкоз – группа заболеваний крови злокачественного характера, которые часто называют еще белокровием или лейкемией. В основе всех болезней лежит замещение нормальных клеток крови, в данном случае, лейкоцитов, их бластами или, по-другому, незрелыми клетками.
Выделяют два типа течения этой патологии – острый и хронический. Причем встречается чаще всего среди всех возможных гемобластозов. У людей с возрастом более 40 лет в диагностике часто выявляется острая миелоидная форма, а у детей это обычно острая лимфобластная форма.
Причины и провоцирующие факторы лейкоза
На сегодняшний день врачи и ученые не нашли ответа на вопрос, по какой причине развивается лейкоз. Однако известно, что в основе заболевания лежит мутация клетки, которая вырабатывает лейкоциты. Это дает начало развитию злокачественного процесса. Мутация этой клетки приводит к тому, что на самом раннем этапе своего развития она не проходит правильную дифференцировку и попадает в кровеносное русло незрелой – она называется бластом.
Однако такие клетки не только появляются в крови, но и происходит их замещение в костном мозге – месте, где осуществляется процесс кроветворения. Все бластные клетки идентичны друг другу, а это говорит о том, что они – клоны одной-единственной клетки подобного типа.
Однако, по какой причине в организме появляется клетка-мутант, неизвестно. Лейкоз крови – одно из самых малоизученных заболеваний, но врачи-гематологи уже сегодня знают о некоторых факторах риска, которые могут привести к такой мутации. Это:
- генетическая предрасположенность, когда в семье есть или был родственник с таким диагнозом – этот показатель в три раза ухудшает как прогноз заболевания, так и риск развития болезни;
- наличие у человека хромосомных аномалий или генетических заболеваний, например, таких патологий, как синдром Дауна, Вискотта-Олдрича, Клайнфельтера;
- воздействие на организм ионизирующей радиации;
- работа с химическими канцерогенами;
- прием цитостатических препаратов.
Часто бывает так, что лейкемия развивается у человека на фоне лечения другого онкологического заболевания – лимфогранулематоза, миеломной болезни, неходжкинских лимфом. Также отмечается связь лейкоза и некоторых вирусных заболеваний – в таком случае он называется вирусным.
Симптомы лейкоза
Острый лейкоз обычно начинается внезапно, на фоне полного здоровья. У пациента появляются повышенная температура тела, симптомы интоксикации, повышенная потливость, отмечаются сильный упадок сил и жизненной энергии, анорексия. Появляются жалобы на постоянные боли в костях, суставах, мышцах.
Часто первые проявления лейкоза могут маскироваться под обычное ОРВИ или даже ангину. Но обращает на себя внимание появление стоматита в его язвенной форме или же развитие гингивита. Если на этой стадии патология не была выявлена, то она постепенно затихает и переходит во вторую. Именно на втором этапе при бессимптомном течении обычно и выявляется острый лейкоз у детей.
В медицине принято различать 4 синдрома, которые типичны для лейкемии:
- анемический проявляется бледностью кожи, головокружением, хронической усталостью, потерей волос, ломкостью ногтей;
- геморрагический отличается увеличением лимфоузлов и миндалин, гепатоспленомегалией, на коже развиваются инфильтраты, поражаются легкие, почки, сердечная мышца;
- интоксикационный проявляется повышенной утомляемостью, головной болью, потерей аппетита, слабостью и вялостью, периодически происходит повышение температуры тела;
- гиперпластический возникает как результат внекостного поражения органов и тканей – происходит их увеличение и нарушение функций, причем особенно часто страдают печень, селезенка, лимфоузлы.
Хронический лейкоз чаще всего выявляется в пожилом возрасте, причем в основном у людей европейского происхождения. Каких-либо специфических симптомов не существует: пациенты жалуются на повышенную потливость, снижение массы тела, происходит увеличение внутренних лимфоузлов, увеличение печени и селезенки на поздних стадиях. Из-за сниженного иммунитета люди с таким заболеванием часто подвержены простудным заболеваниям и пневмониям.
Признаки лейкоза могут быть выражены ярко, а могут никак себя не проявлять. К тому же часто они маскируются под другие болезни. Поэтому любое заболевание должно рассматриваться как потенциально опасное для развития этого онкологического заболевания крови.
Классификация лейкоза
Все лейкозы делятся на острые и хронические. Однако надо понимать, что острый лейкоз никогда не станет хроническим, а хронический не может обостриться – в этом отличие от других заболеваний. Данная терминология используется только для удобства в медицине.
В зависимости от того, какие клетки крови оказываются пораженными, лейкемия бывает лимфобластной, миелобластной, монобластной, миеломонобластной и других видов. Хронические лейкозы, которые в основном встречаются у взрослых, представлены более широко.
Лейкоз протекает в три стадии. На первой появляются первичные симптомы общего плана, которые характерны практически для любого заболевания. Она носит название моноклоновая, протекает долго и относительно доброкачественно.
На второй стадии симптомы лейкоза проявляются очень ярко и полностью соответствуют гематологическим проявлениям гемобластоза. Начинается формирование вторичных клонов опухоли, и часто его называют бластным кризом.
Третья стадия – терминальная, здесь практически полностью прекращается формирование новых клеток крови. Она наступает в случае отсутствия правильной диагностики и лечения на первых двух этапах.
Осложнения лейкоза
Лейкемия – серьезное и очень опасное заболевание. Без лечения оно может вызвать массу различных осложнений. Так, например, это может быть аутоиммунный криз, который протекает с сильной слабостью, лихорадкой, повышенной температурой тела, болями в животе. Синдром сдавливания средостения вызывает сильную одышку, воротной вены – асцит, кишечника – его непроходимость.
Может отмечаться поражение селезенки, что вызывает ее инфаркт из-за нарушения кровообращения. Смерть пациента может случиться из-за кровоизлияния в мозг, желудочно-кишечного кровотечения, пневмонии, сепсиса, почечно-печеночной недостаточности.
Диагностика лейкоза
Для диагностики используются лабораторные анализы. Они помогают выявить заболевание на любой стадии его развития:
- Общий анализ крови с четким определением лейкоцитов по их разновидностям. Это будет лучшим решением для первичной диагностики заболевания.
- Биохимия крови помогает выявить нарушения, которые могут возникнуть в почках или печени.
- Биопсия костного мозга помогает выяснить, насколько сильно его поражение, поставить точный диагноз и определить тактику лечения пациента.
- Гистология тканей костного мозга позволяет определить количество злокачественных клеток и их характер.
- Молекулярно-генетический анализ делается, когда диагноз находится под сомнением.
- УЗИ органов брюшной полости помогает понять, насколько поражены внутренние органы.
- Рентгенография грудной клетки помогает выявить увеличенные лимфоузлы и другие патологические процессы, протекающие в легких.
Выявлением и терапией занимается врач-гематолог, однако заподозрить заболевание у детей может педиатр, а у взрослых – терапевт.
Лечение лейкоза
Терапия проводится в условиях стационара. Основной метод – химиотерапия. Это метод, при котором пациент принимает специальные препараты, уничтожающие злокачественные клетки. Схемы подбираются строго индивидуально.
Для улучшения состояния крови выполняется введение эритроцитарных масс, тромбоцитов, изотонических растворов. При присоединении инфекции необходима антибактериальная терапия. Лучевое лечение используется перед тем, как пересадить костный мозг: такая пересадка выполняется при отсутствии эффекта от других методов лечения.
Если организм пожилого человека с лейкозом не в состоянии вынести тяжелого лечения, назначается паллиативная терапия.
Рекомендации при лейкозе даются каждому пациенту индивидуально. Все лечение проводится в стерильных боксах, чтобы пациент не смог подхватить инфекцию, которая может оказаться для него смертельной.
Прогноз при лейкозе благоприятен в том случае, если есть положительный результат от лечения. Однако иногда есть риск развития рецидива, и тогда вылечить пациента уже очень сложно.

терапевт, гематолог, КМН, консультации онлайн
опыт работы 10 лет
отзывы оставить отзыв
Клиника
м. Красные Ворота
м. Сухаревская
Записаться на прием
Отзывы
Специалист и мастер своего дела, подходит к консультациям с юмором и серьезностью одновременно. Даже при температуре и с плохим состоянием здоровья (во время эпидемии гриппа) успокоила и рассказала что делать и как не паниковать. Спасибо большое!
Марина
11.02.2020 17:40:42
очень, очень скептически относилась к озонотерапии, но все-таки решилась на курс. И эффект от процедур однозначно есть. спасибо, «Поликлиника.ру»!
Татьяна Виленовна — отличный врач и внимательный человек. Я очень благодарна за консультации и последующее наблюдение.
Хочу выразить благодарность Татьяне Виленовне за отзывчивость, профессионализм, юмор, внимательность! Татьяна Виленовна в моем представлении — такая, какими мы хотим видеть всех врачей! Спасибо!
Услуги
- Название
- Прием (осмотр, консультация) врача-гематолога первичный2300
- Прием (осмотр, консультация) врача-гематолога повторный1900
Статьи о здоровье
Другие специалисты
Лейкемия у детей: как снизить риск опасной болезни?
![]()
![]()

Лейкемия, которую многие знают как лейкоз или «белокровие» – опаснейшая болезнь. Это самый распространенный тип рака у детей в возрасте до 15 лет, и ежегодно около 5 тысяч детей в стране начинают страдать от лейкемии. Это генетическое заболевание, но на его возникновение влияют различные факторы. Некоторыми мы можем управлять, а значит, защитить ребёнка от развития рака крови. MedAboutMe рассказывает обо всех опасностях.
Лейкемия по наследству?

Лейкемия – это генетическая болезнь. Она связана с ДНК, той самой последовательностью, в которой зашифрована вся наша генетическая информация. Именно коды ДНК определяют, как будут развиваться, расти и функционировать клетки организма.
Лейкоз развивается вследствие мутаций в ДНК, провоцирующих патологические изменения костного мозга на клеточном уровне. Клетки крови, которые продуцируются костным мозгом, становятся неправильными, аномальными. Они мешают появляться и выполнять свою работу новым здоровым клеткам. Красных кровяных телец становится мало, аномальных белых много, отсюда – название лейкемии – белокровие.
Эти мутации редко наблюдаются среди родственников. Генетические мутации, связанные с лейкозом, обычно развиваются после зачатия, а не наследуются от генов родителей. Ребёнок может унаследовать изменения ДНК от своих родителей, или они спонтанно образуются уже после слияния половых клеток отца и матери.
Но хотя лейкоз не относится к наследственным заболеваниям, есть определенные генетические аномалии, увеличивающие вероятность развития лейкемии. И некоторые такие триггерные мутации могут передаваться от родителей к детям.

Исключение – семейный острый миелоидный лейкоз, редкая наследственная форма лейкемии.
Что повышает риск белокровия?
Есть триггеры заболевания, на которые мы имеем возможность влиять, и есть те, которые можно только учитывать. Знать надо обе группы – чтобы снизить риск или чтобы принять профилактические меры и вовремя заметить симптомы.
Генетика как фактор риска детского лейкоза
Ребёнок может наследовать генетические аномалии, повышающие риск. Мутации также могут появляться спонтанно – по неизвестным причинам или под воздействием внешних факторов.
Если у ребёнка есть родственник первой степени, родитель или брат/сестра с лейкозом, это фактор риска хронического лимфолейкоза. В 2019 году было опубликовано исследование, в котором выявили, что такие мутации, как FLT3-ITD и NRAS, чаще регистрируются у тех людей, у которых есть AML-M5, тип AML, образующийся в незрелых лейкоцитах. То есть была выявлена связь между определенной генетической последовательностью и опасностью развития лейкоза.
Следующие наследственные генетические заболевания также способны увеличить риск всех форм лейкемии:
- синдромы Дауна, Блума, Клайнфельтера, Ли-Фраумени;
- атаксия, телеангиэктазия;
- нейрофиброматоз;
- малокровие Фанкони.
Возраст и пол
От такой формы лейкемии, как острый лимфоцитарный лейкоз, намного чаще страдают дети до 15 лет. У мальчиков лейкемия встречается чаще, чем у девочек.
Воздействие окружающей среды
Окружающая среда, влияние химических веществ и нездоровые привычки способствуют генетическим мутациям. Это мутагенные факторы, которые способны вызывать аномалии ДНК или провоцировать манифестацию болезни при наличии склонности. Образ жизни также может увеличить риск. К таким триггерам, «спусковым крючкам» болезни относятся воздействие токсических соединений, сигаретный дым в том помещении, где находится ребёнок, и курение матери во время беременности.
Промышленные химпрепараты, а также избыток бытовой химии вокруг ребёнка могут стать триггерами заболевания.
Бензол – одно из самых опасных соединений. Это химическое вещество присутствует во многих товарах вокруг нас, от клея и чистящих средств до моющих жидкостей и красок.
Общие симптомы детского лейкоза

Если у ребёнка есть любой из следующих симптомов, необходимо обратиться к врачу.
- Анемия: возникает, когда в организме наблюдается дефицит эритроцитов. Красные кровяные тельца ответственны за перенос кислорода по всему телу, и их нехватка вызывает усталость, слабость, одышку, головную боль, бледность кожи, мерзлявость и головокружения.
- Частые инфекционные заболевания: при лейкозе в крови выявляется повышенное количество лейкоцитов, но большинство из них аномальные, они работают неправильно и не защищают организм от инфекции.
- Синяки, гематомы и кровотечения: недостаток тромбоцитов вызывает длительные кровотечения и легко возникающие синяки.
- Костные, суставные боли: при лейкозе аномальные кровяные тельца могут накапливаться в тканях суставов или рядом с костной поверхностью и вызывать болевые ощущения.
- Припухлости и отеки: отечность разной локализации и припухлость лимфатических узлов могут быть признаком лейкемии. Отек может поражать различные части тела, от брюшной полости до лица, рук, шеи и т. д.
- Плохой аппетит, боль в области живота, похудение: если лейкозные клетки вызвали отек печени, почек или селезенки, отечность давит на окружающие органы, в том числе на желудок. В результате падает аппетит, растет дискомфорт, ребёнок худеет.
- Кашель или затруднение дыхания: лейкоз способен поражать ткани грудной клетки, из-за отечности возникает давление на трахею, дыхательные пути сдавливаются.
- Головная боль, приступы рвоты и судорог возникают из-за поражения головного или спинного мозга лейкозными клетками. Могут развиваться трудности с концентрацией внимания, проблемы с балансом, затуманенное зрение.
- Кожная сыпь: лейкозные клетки привести к появлению мелких, темных пятен (хлорома или гранулоцитарная саркома). Еще один вид кожных проявлений – петехии, это крошечные пятна подкожных кровоизлияний.
Профилактика лейкемии
Хотя у нас нет контроля над определенными факторами риска, такими как возраст и пол, но можно снизить риск лейкемии. Что может помочь:
- изучение семейной истории;
- отказ от курения всей семьей;
- избегание контакта с бензолом, формальдегидом и другими токсичными химическими веществами;
- поддержание здоровой массы тела путем регулярных физических упражнений и здорового питания для ребёнка и взрослых;
- регулярные профилактические осмотры у всех специалистов.
Читайте далее

Тревожность и как с ней бороться
Тревогу может испытывать любой человек в связи со стрессом, неприятностями. Но бывает, что, вроде, беспокоиться не о чем, а тревога мучает всё равно.
Опубликовано 02.03.2020 10:15, обновлено 17.04.2020 18:00
Использованные источники
Neonatal leukaemia. / Roberts I, Fordham NJ, Rao A, Bain BJ. // Br J Haematol – 2018 Jul – 182(2)
The role of inflammation in leukaemia. / Giles FJ, Krawczyk J, O’Dwyer M, et al. // Adv Exp Med Biol – 2014
Mutation topography and risk stratification for de novo acute myeloid leukaemia with normal cytogenetics and no nucleophosmin 1 (NPM1) mutation or Fms-like tyrosine kinase 3 internal tandem duplication (FLT3-ITD). / Zhou YL, Wu LX, Peter Gale R, et al. // Br J Haematol – 2020 Feb 26
Читайте также
Как распознать лейкоз по анализу крови?
Врач-гематолог, доктор медицинских наук Сергей Семочкин про острый лимфобластный лейкоз

Врач-гематолог, профессор кафедры онкологии, гематологии и лучевой терапии РНИМУ им. Н. И. Пирогова Минздрава России, доктор медицинских наук Сергей Семочкин рассказал: можно ли распознать острый лимфобластный лейкоз (ОЛЛ) на ранней стадии и поставить диагноз по анализу крови; объяснил, как лечат ОЛЛ и кому показана трансплантация костного мозга (ТКМ).
Каковы ранние симптомы острого лимфобластного лейкоза? Можно ли их увидеть и распознать ОЛЛ?
В данном случае все достаточно просто, потому что слово «острый» означает, что заболевание внезапное и зачастую симптомы очень выразительные. Самый частый симптом — это лихорадка, т.е. повышение температуры тела. Лихорадка может быть как субфебрильной, так и ярко выраженной, до 39 градусов. Появятся изменения, связанные с поражением костного мозга. Снижение гемоглобина приведет к слабости и быстрой утомляемости. Могут увеличиться лимфатические узлы, появиться дискомфорт в брюшной полости за счет того, что увеличиваются размеры печени и селезенки. Могут быть проявления кровоточивости — даже во время чистки зубов. У некоторых пациентов ОЛЛ может начаться с неврологических проявлений – головных болей, головокружения и прочих проблем. Симптоматика обширная, но в данном случае она является достаточно острой, внезапно возникшей.
Смотрите видео на нашем сайте.
Можно ли поставить диагноз по анализу крови? Что он покажет?
Как правило, в анализе крови есть ярко выраженные показатели: изменены ростки кроветворения, количество лейкоцитов выходит за пределы нормы — может упасть ниже нормальных значений, а может стать запредельно огромным. Мне встречались пациенты, у которых количество лейкоцитов при норме от 4 до 9 тысяч повышалось до 200 тысяч на мкл. Тромбоциты тоже в ряде случаев очень сильно снижены, но главное – изменение количества лейкоцитов. Очень важным маркером является выход опухолевых клеток в кровь, когда в крови появляются незрелые ранние клетки, которые называют бластными. Если в анализе крови выявили бластные клетки, то это, скорее всего, либо острый лейкоз, либо миелодиспластический синдром.
Как пациент попадает к гематологу?
Анализ крови с характерными изменениями — повод для немедленного вызова скорой помощи и госпитализации пациента в профильный стационар. При лечении детей и подростков у онкогематолога, как правило, есть один-два дня ни диагностику, лечение необходимо начинать, как можно раньше. В диагностику входит повторный анализ крови, затем – верификация диагноза, для которой проводят биопсию костного мозга. У маленьких детей ее проводят под общей анестезией, у взрослых — под местной. С помощь небольшой иглы делаю прокол грудины или подвздошной кости. У детей пункцию грудины не делают. Полученный образец костного мозга, который выглядит как обычная пробирка с кровью, отправят в лабораторию, где для подтверждения диагноза проведут целый спектр исследований. Главный критерий – увеличение количества бластных клеток. Только по внешнему виду и по количеству бластных клеток определить вариант лейкоза – невозможно. Еще в 1913 году установили, что есть лимфоидный, а есть миелоидный вариант лейкоза. Для верификации применяются специальные лабораторные методы: иммунологические и химические. Существует специальный прибор – проточный цитометр, с помощью которого определяют маркеры, характеризующие данный тип клеток. Для определения подвида острого лейкоза, применяют целый спектр генетических исследований, чтобы выйти на более целевую терапию у этих пациентов.
Каковы причины возникновения ОЛЛ? Существует мнение, что этот вид лейкоза очень сильно взаимосвязан с экологическими проблемами, передается по наследству и часто возникает у тех, кто уже переболел каким-то онкологическим заболеванием. Правда это или нет?
Истинную причину возникновения лейкоза у взрослых можно выявить только в 5% случаев, в 95% совершенно непонятно, что там к чему привело. У детей все несколько интересней.
Как возникает лейкоз? В генетическом материале клетки возникает некая первичная мутация, которая сама по себе далеко не всегда приводит к лейкозу. В дальнейшем к этой мутации присоединяются другие, и когда болезнь все же возникает, в клетке накоплено уже много молекулярных событий, сочетание которых привело возникновению заболевания. Пик острого лимфобластного лейкоза приходится на детей от двух до четырех лет, потом заболеваемость падает. Следующий пик приходится на 18-29 лет, потом снова спад. После 60 лет — опять небольшой рост.
У части детей раннего возраста прослеживается некая врожденная составляющая этой проблемы. Встречаются случаи ОЛЛ у плода или новорожденного, когда ребенок рождается уже с заболеванием, либо заболевает в течение первого года жизни. Исследования пуповинной крови показали, что у новорожденных встречаются лейкемические поломки, врожденные мутации, которые могут привести к возникновению лейкоза. И мутацию эту вызывает наследственный фактор, сработавший во время внутриутробного развития. По разным данным, общее количество таких младенцев составляет от 1 до 5%. Дальше многое зависит от инфекционной обстановки, сложившейся вокруг ребенка. Многочисленные инфекции, перенесенные в детстве, способствуют формированию нормальной иммунной системы, которая нейтрализует наследственный фактор.
Если говорить про экологические проблемы, то с ними четкой связи не выявлено.
Влияет ли УФ-излучение, СВЧ, солнечные лучи, радиация?
В Хиросиме и Нагасаки повышенная заболеваемость держалась около 12 лет. После Чернобыля у многих пострадала щитовидная железа, но заболеваемость лейкозами не выросла. Все зависит от типа изотопов попавших в окружающую среду. В Фукусиме тоже этого не случилось, потому что концентрация радиоактивных веществ сильно разбавилась морской водой.
Вред ультрафиолета научно доказан только в отношении меланомы. Четкой связи с ОЛЛ нет. Своим бывшим пациентам мы не разрешаем посещать солярий и не рекомендуем загорать, потому что хотя связь и не доказана, совсем исключать этот фактор тоже нельзя.
Если говорить об СВЧ-излучении, домашние микроволновые печи абсолютно безопасны.
Как лечат ОЛЛ? Что ждет пациента?
Концепция лечения ОЛЛ, которая до сих поре лежит в основе протоколов лечения ОЛЛ, была разработана американским педиатром Дональдом Пинкелем еще в 1962 году. Она включает в себя четыре этапа: индукция ремиссии, консолидация, воздействие на центральную нервную систему и длительный этап поддерживающей терапии на протяжении двух-трех лет. Во всем мире проводится лечение по клиническим протоколам, разработанным в результате кооперированных исследований. Согласно некоторым работам, строгое следование протоколам повышает выживаемость пациентов на 15-20% по сравнению с индивидуализированным лечением. В протоколе прописаны все действия: от первого дня до последнего. В нем есть указания, как и в какой момент оценивать возникающие осложнение и что с ними делать. В России два центра, активно ведущих такие протоколы. Центр им. Дмитрия Рогачева, где Александр Исаакович Карачунский в течение многих лет, с начала 1990-х, ведет серию протоколов «Москва — Берлин». Каждые пять лет дизайн протоколов пересматривают, чтобы улучшить лечение отдельных категорий пациентов. уже в течение многих лет с начала 90-х годов серию протоколов Москва-Берлин. Каждые пять лет меняется дизайн протоколов, направленных на улучшение лечения отдельных категорий пациентов. Во взрослой практике — это НМИЦ гематологии, где ведут кооперированные исследования по острому лимфобластному лейкозу у взрослых.
В каких случаях показана трансплантация костного мозга (ТКМ)?
В отличие от острого миелоидного лейкоза, показаний к аллогенной (от донора) ТКМ меньше. Ее назначают пациентам, которые не достигли ремиссии в указанные протоколом сроки или имеют неблагоприятный цитогенетический вариант заболевания. В детской практике выздоравливают более 90% детей, и примерно 15-20% являются кандидатами для аллогенной ТКМ. У взрослых процент пациентов нуждающихся в трансплантации несколько выше, за счет того, что генетических операций высокого риска становится намного больше и ответ на стандартное лечение хуже. Когда мы обсуждали хронический миелолейкоз, там фигурировала филадельфийская хромосома — транслокация (9;22). При ОЛЛ это абсолютно негативный фактор прогноза. У детей такая мутация встречается меньше чем в 5% случаев, у людей старше 50-60 лет примерно половина В-линейных ОЛЛ будет с филадельфийской хромосомой. В отличие от хронического миелолейкоза, применение ингибиторов тирозинкиназы при остром лимфобластном лейкозе не столь успешно. Вот поэтому во взрослой практике ТКМ необходимо проводить примерно 30% пациентов. Возрастной порог для аллогенной ТКМ — в районе 55 лет, это разумно.
Как часто случаются рецидивы с ОЛЛ?
Если мы говорим про взрослых людей, то рецидивы случаются почти в 40% случаев. Бывают ранние рецидивы, которые случаются прямо на терапии. В таком случае необходимо менять лечение, делать его более интенсивным и тяжелым. В таких случаях, как правило, показана ТКМ. Поздний рецидив может случиться и через 20 лет. К сожалению мы не можем убрать причину, которая вызывает это заболевание — оно может вернуться.
Можно ли планировать беременность после ОЛЛ?
Длительная химиотерапия нарушает фертильность, поэтому лучше провести криоконсервацию спермы/яйцеклетки, а еще лучше эмбриона — это более надежный способ. У мужчин, как правило, серьезно нарушается сперматогенез, но у женщин дело обстоит несколько лучше. Вероятность забеременеть и выносить здорового ребенка высока. Если прошло не менее пяти лет в ремиссии, никаких ограничений нет.
Может ли беременность быть провоцирующим фактором для рецидива?
Скорее, нет. Это не такое частое явление, как при некоторых других заболеваниях, где беременность действительно может стать провоцирующим фактором.
Передается ли ОЛЛ по наследству?
Лимфобластный лейкоз – редкое заболевание, поэтому вероятность того, что он случится у ребенка, рожденного от родителей после ОЛЛ, крайне мала.
Как будут лечить ОЛЛ в будущем?
Представляется, что в основе лечения онкологических заболеваний в будущем станет активация собственного иммунитета. Нам необходимо настроить иммунную систему таким образом, чтобы она распознавала и убирала раковые клетки. Сейчас мы находимся на раннем этапе развития CAR-T-терапии, но через какое-то время технологии настолько усовершенствуются, что, скорее всего, она станет одним из основных методов терапии при целом ряде онкогематологических заболеваний. Суть метода заключается в том, что у пациента собирают его собственные Т-лимфоциты и отправляют в специальную лабораторию. Эта лаборатория может быть в другом городе, стране — не важно. В лаборатории эти Т-лимфоциты перепрограммируются: в них появляется информация об опухолевых клетках, присутствующих в организме пациента. После перепрограммирования Т-лимфоциты вводят обратно пациенту, она находят раковые клетки и возникает ремиссия. Основные проблемы – создать качественный процесс распознавания и разработать стандартные протоколы лечения.
Много вопросов возникает в понимания биологии заболевания, потому как каждый конкретный случай весьма индивидуален. Мы знакомы только с грубыми поломками, но каждая отдельная поломка провоцирует различное течение болезни. Мы уже сейчас можем полностью секвенировать геном опухолевой клетки и главное научиться понимать, что в патогенезе является ключевым и как на это можно воздействовать, тогда мы ближе подойдем к полному излечению болезни. За этим будущее.